Background and Objective: Intraocular lymphoma (IOL) is a heterogenous category of rare malignancies that are often misdiagnosed and underrecognized. The rarity of IOL impedes clinical research and contributes to difficulty in standardizing its management. In this article we review the existing scientific literature to identify the current diagnostic tools and discuss comprehensive management of various categories of IOL. Our objective is to increase disease recognition of IOL as a whole and explore updated management options for each subtype.
Methods: PubMed and Embase were searched for publications using the terms ‘intraocular lymphoma’, ‘vitreoretinal lymphoma’, ‘uveal lymphoma’, ‘iris lymphoma’, ‘choroidal lymphoma’ and ‘ciliary body lymphoma’ published from 1990 to June 2021. Inclusion criteria were English language articles. Exclusion criteria were non-English language articles, case reports and animal studies.
Key Content and Findings: IOL often presents in middle-aged and older patients with symptoms of floaters and vision changes, but a broad array of clinical signs and symptoms are possible depending upon subtype. IOL can be subdivided by location of involvement into vitreoretinal and uveal lymphoma. These subtypes express key differences in their pathophysiology, clinical presentation, histology, prognosis, and treatment. Primary vitreoretinal lymphomas (PVRL) generally originate from B-lymphocytes and are associated with central nervous system (CNS) lymphoma. Ophthalmic findings include retinal pigment epithelium changes with yellow subretinal deposits known as “leopard spotting.” Primary uveal lymphomas generally originate from low-grade B-lymphocytes invading the choroid and carry an improved prognosis compared to vitreoretinal lymphomas. Funduscopic findings of primary uveal lymphoma include yellow to pink-yellow choroidal swelling with infiltrative subconjunctival “salmon-patch” lesions. Diagnosis for IOL is often delayed due to insidious onset, low prevalence, and tendency to mimic diseases such as uveitis. Diagnosis may be challenging, often relying on biopsy with specialized laboratory testing for confirmation of IOL. Optimal treatment regimens are currently debated among experts. Management of IOL is best coordinated in association with neuro-oncology clinicians due to the tendency for intracranial involvement.
Conclusions: IOL represents a group of multiple malignancies with distinct clinicopathologic features. Future outlook for treatment and prognosis of IOL is likely to improve with less invasive molecular diagnostic techniques and increased awareness. Clinicians should be circumspect in all patients with possible IOL and promptly refer to oncologic specialists for rapid evaluation and treatment.
Intraocular lymphoma (IOL) is a heterogenous category of rare malignancies that are often misdiagnosed and underrecognized. The rarity of IOL impedes clinical research and contributes to difficulty in standardizing its management. In this article we review the existing scientific literature to identify the current diagnostic tools and discuss comprehensive management of various categories of IOL.
IOL may be categorized depending upon the intraocular structures affected, lymphocytic origin from B- or T-lymphocytes, and whether disease is confined to the central nervous system (CNS) (primary IOL) or metastatic from systemic lymphoma (secondary IOL). When classified by intraocular location, it is divided into vitreoretinal and uveal lymphoma (1-4). Uveal lymphoma is that which affects the iris, ciliary body and/or choroid while vitreoretinal lymphoma affects the retina and/or vitreous. Vitreoretinal lymphomas and uveal lymphomas represent two generally distinct disease processes and will be discussed separately. Primary vitreoretinal lymphoma (PVRL) is associated with development of CNS lymphoma before, after or concurrent with diagnosis (5). It should be suspected in immunosuppressed patients of advanced age presenting with vision changes and floaters which do not respond to steroids (5,6). Uveal lymphoma is unrelated to CNS lymphoma yet arises in patients of advanced age with blurred vision or metamorphopsia with a predilection for males (7,8). The clinical presentation of all types of IOL may be difficult to distinguish from uveitis, degenerative changes, and other ocular diseases and for these reasons diagnosis is often delayed (1,9-11). We review the current literature regarding IOLs and their management to increase disease recognition of this important entity. We present the following article in accordance with the narrative Narrative Review checklist (available at https://aes.amegroups.com/article/view/10.21037/aes-21-59/rc).
Online databases PubMed and Embase were searched for publications using the terms ‘intraocular lymphoma’, ‘vitreoretinal lymphoma’, ‘uveal lymphoma’, ‘iris lymphoma’, ‘choroidal lymphoma’, and ‘ciliary body lymphoma’ published from 1990 to June 2021. Inclusion criteria were English language articles which discussed lymphomas involving intraocular tissue, clinical research or nonclinical, meta-analyses, reviews, or case series. Exclusion criteria were non-English language articles, case reports and animal studies (Table 1).
| Items | Specification |
|---|---|
| Date of search | May 2021 |
| Databases and other sources searched | PubMed, Embase |
| Search terms used | Intraocular lymphoma, vitreoretinal lymphoma, uveal lymphoma, iris lymphoma, choroidal lymphoma, ciliary body lymphoma |
| Inclusion and exclusion criteria | Inclusion criteria: English language, clinical or nonclinical meta-analyses, reviews, or case series |
| Exclusion criteria: non-English language articles, case reports, animal studies | |
| Selection process | Selection was conducted by authors independently |
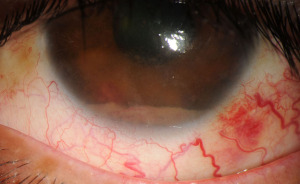
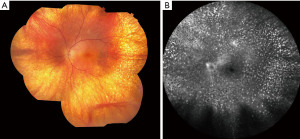
PVRL is defined by monoclonal neoplastic lymphocytes detected within the vitreous and/or retina without evidence of systemic metastasis or disease outside of the CNS (Figure 1A-1C) (2,12). Though it is the most common form of IOL, it is still exceedingly rare. Estimates of its incidence center around 0.046 per 100,000 person-years (13). However, incidence of PVRL is thought to be rising in parallel with increasing incidence of primary central nervous system lymphoma (PCNSL) (6,14). PVRL is intimately associated with PCNSL and considered to fall under the category of PCNSL. A minority of vitreoretinal lymphomas are not primary, i.e., they are the result of disseminated systemic lymphoma. Many of these cases however (particularly the diffuse large B-cell subtype) involve the uvea and eventually also develop CNS disease (5,15). Given the rarity of PVRL and paucity of randomized clinical trials, knowledge of its risk factors and epidemiology have mostly been gleaned through research of PCNSL (16). PVRL appears to affect both genders equally and commonly arises in the 6th decade of life (1). Immunodeficiency and immunosuppression are correlated with an elevated risk of PVRL. PVRL is most often bilateral, although asymmetric involvement is possible (7,17). The malignant cells of PVRL are typically B-lymphocytes, histologically categorized as large B-lymphocytes. A very small minority of vitreoretinal lymphomas originate from T- or NK/T-cells. Cases of vitreoretinal lymphoma derived from T-lymphocytes are usually associated with metastatic spread from systemic adult T-cell leukemia/lymphoma and are therefore secondary IOL, but very rarely PVRL of T-cell origin has been reported (Figure 2) (1,2). Some cases of T-cell lymphoma which are metastatic from a non-CNS site will also develop CNS disease. The most common presenting symptoms are blurred or decreased vision and floaters (18). PVRL may “masquerade” as multiple other ocular diseases such as infectious uveitis, noninfectious uveitis or degenerative changes, which further complicates diagnosis (5,6,17). Steroids are often prescribed and typically result in no improvement, though occasionally symptoms such as floaters may temporarily improve (5). It is important to remember that PCNSL may be diagnosed prior to PVRL, be detected simultaneously, or develop after diagnosis. Adults with uveitis and neurologic symptoms should be suspected of possible PVRL. PVRL is generally an aggressive malignancy and prognostic factors are not well defined, but presence of concurrent PCNSL is generally associated with poorer prognosis (5,19,20). More than half of PVRL patients (some estimate up to 92%) either have concurrent PCNSL at diagnosis or will eventually develop it, which makes monitoring for CNS disease critical (2,5,13). Early recognition and prompt diagnosis are essential for PVRL and all types of IOL.
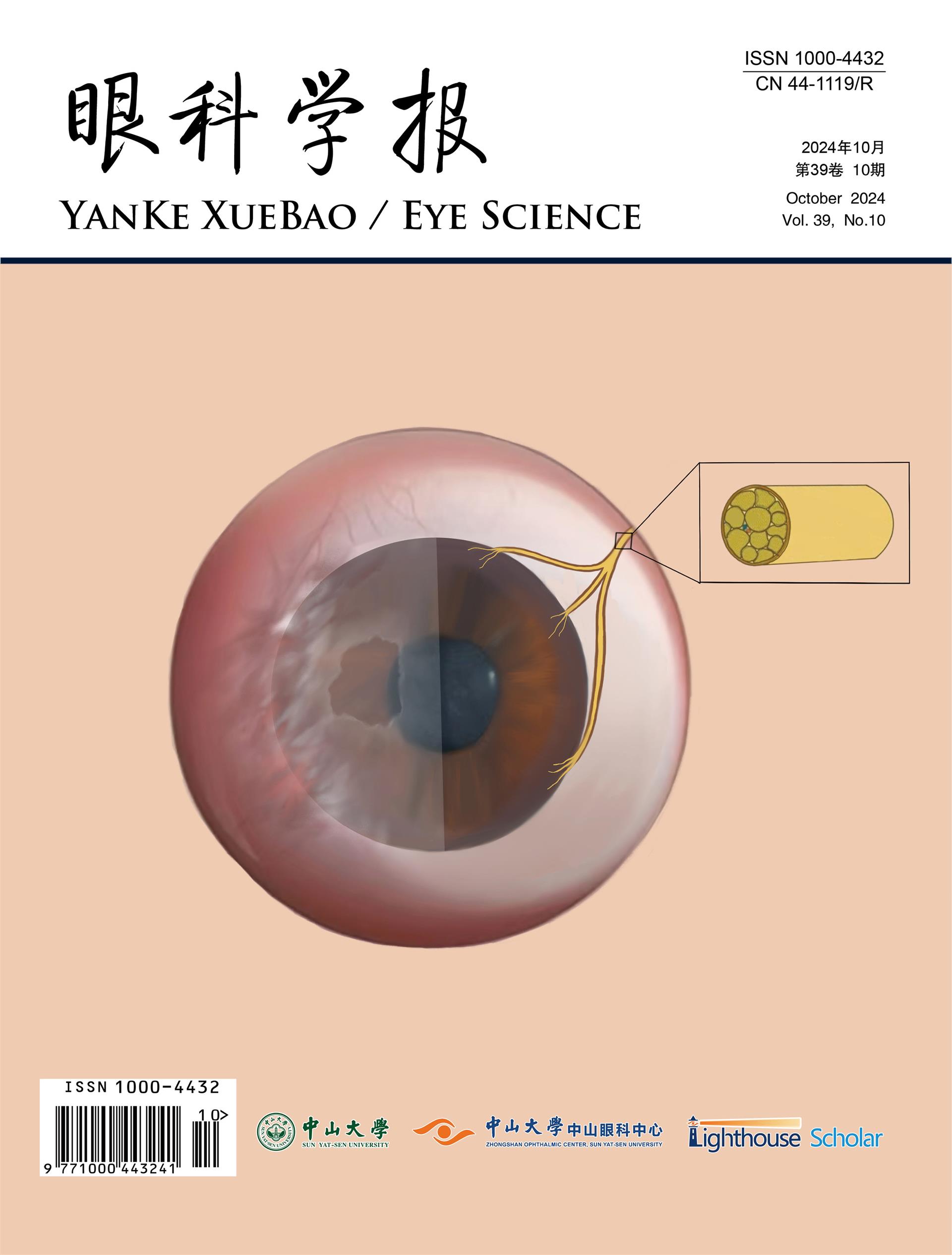
Vitreous haze and vitreous cells are the primary findings in patients with PVRL, and may appear as large clumps, sheets, or as large homogenous single cells. A highly characteristic feature of PVRL is retinal pigment epithelium (RPE) changes with yellow subretinal deposits, which may lead to RPE detachments (1,2,15,21). The yellow subretinal deposits with overlying RPE changes have been described as “leopard spotting”. This is often better visualized on fluorescein angiography as hyper- and hypofluorescent lesions (Figure 3) (5,16). Additional findings which may be seen on FA include granularity, blockage, and hypofluorescent focal lesions in early and late stages with involvement of the outer retina (2,15). Lymphoma cells often create focal hyperreflective nodularity of the retinal pigmental epithelium on optical coherence tomography (OCT) (Figure 4) (2,16). The anterior segment is usually quiet and without obvious pathology in PVRL or may show mild inflammation. Involvement of the iris may be seen in T-lymphocyte mediated vitreoretinal lymphoma however, which more commonly presents with anterior uveitis (1,22). The absence of cystoid macular edema in PVRL is a distinguishing feature from uveitis, where it is often present (2,5); however, PVRL can in rare instances demonstrate cystoid macular edema and/or serous retinal detachment.

PVRL has generally been diagnosed via isolation of malignant lymphocytes from the vitreous. Unfortunately, this is an invasive and technically challenging task. Diagnostic yield is a crucial consideration; malignant cells are often difficult to isolate from the vitreous, overshadowed by reactive inflammatory cells in the specimen and fragile to necrosis (1,23). Pars plana vitrectomy has shown to have an improved diagnostic yield in comparison to vitreous aspiration and is currently the most commonly used technique. It may also offer some improvement in patient visual acuity and symptoms by removing debris (2,6,13). Pathologists should be consulted prior to vitrectomy in order to plan for ideal technique and transport to lab for processing within one hour; fetal calf serum and cold transport have been shown to improve yield (1). Recent steroid treatments can negatively impact detection of malignant cells, so diagnostic vitrectomy should not be performed within two weeks of steroid treatment (2). Due to these challenges, false negatives are relatively common (up to 30%) and patients may require multiple biopsies or undergo chorioretinal biopsy in order to identify malignant lymphocytes for definitive diagnosis (2,5,24).
In recent years molecular diagnostic tools have become useful adjuncts for diagnosis. Neoplastic monoclonal B- or T-lymphocytes in vitreous fluid can be identified via flow cytometry, immunohistochemical staining and/or polymerase chain reaction (PCR) when the vitreous specimen is sufficient to allow it. Neoplastic lymphocytes can also be detected by skewed gamma to kappa light chain ratio, abnormal immunoglobulin heavy chain gene rearrangements, or T-cell receptor gene rearrangements in vitreous cell populations (12,13). Cytokine analysis of aqueous or vitreous fluid interleukins has been shown to be relatively sensitive and specific (1,25). IL-10/IL-6 ratio >1.0 is indicative of lymphoma, while a ratio <1.0 is more suggestive of uveitis. Sensitivity has been reported to be above 88% (25,26). However, these molecular signals can sometimes be detected in reactive inflammatory cells and do not always indicate lymphoma. PCR detection of MYD88 gene mutation has been shown to improve diagnostic certainty, as this gene is frequently mutated in PVRL and PCNSL cell populations. Presence of MYD88 gene mutation increased the sensitivity of vitreous fluid analysis from 62% to 90.5% and improved the negative predictive value from 85.5% to 96% in one study (27). The sensitivity and specificity of each molecular diagnostic tool is difficult to define, given the low number of clinical trials and generally small patient populations in each. It is most useful to combine histopathologic, molecular and radiologic methods in order to increase diagnostic certainty (5,16).
Detection of altered microRNA expression in bodily fluids has recently been studied as a promising less invasive diagnostic tool for PVRL and a variety of other malignancies (11,28,29). Upregulation of specific microRNAs was demonstrated in the cerebrospinal fluid (CSF) of CNS lymphoma patients, and subsequently many alterations in microRNA expression have been found in the vitreous and serum of patients with vitreoretinal lymphoma (11). Tuo et al. demonstrated significantly elevated microRNA-155 in the vitreous of patients with uveitis compared to patients with PVRL, offering a potential diagnostic tool for distinguishing these two illnesses with often overlapping clinical presentation (11). A more recent study has shown serum microRNA-6513-3p levels can successfully discriminate vitreoretinal lymphoma from uveitis, while serum microRNA-326 levels can successfully distinguish vitreoretinal lymphoma from macular hole and/or epiretinal membrane (29). While existing studies are limited in sample size and therefore generalizability, microRNA expression as a diagnostic tool for PVRL is a promising frontier that is likely to expand diagnostic options and may reduce the need for invasive diagnostic vitrectomy in the future (11,28,29).
Imaging should always be included in diagnostic workup, particularly due to the propensity for patients to have concomitant or develop subsequent PCNSL. Magnetic resonance imaging (MRI) with contrast is the preferred modality for detection of CNS lesions (2,5,13). Lumbar puncture for analysis of CSF and/or brain biopsy may be indicated. Histopathologic detection of malignant lymphocytes in CSF or brain tissue is considered diagnostic for PVRL/PCNSL in this case and can avoid the need for vitrectomy or invasive chorioretinal biopsy (13,15).
Chorioretinal biopsy is a complex procedure which can be utilized in patient cases without a clear diagnosis following thorough patient history, systemic workup and conventional diagnostic methods. It may be indicated for patients with high remaining suspicion of PVRL despite inconclusive diagnostic vitrectomy results, patients with disease unresponsive to treatment, and patients whose disease may not involve the vitreous (23). It has traditionally been challenging to perform but has become more commonplace over time with the increased availability and use of small-gauge instruments (23,24). Chorioretinal biopsy may be performed via an external approach or transvitreal (23). Choice of biopsy site is an important consideration. The ideal chorioretinal biopsy site from a histopathologic standpoint is one situated on the border of abnormal tissue and adjacent normal tissue, allowing for direct comparison of both tissue types in one specimen. Vessels involved in the biopsy site must be cauterized to achieve hemostasis however, and the effects of distal blood supply disruption should be considered when selecting a biopsy site as well as the risk of future biopsy site fibrosis and proliferative vitreoretinopathy (24). Excision of the chorioretinal tissue should aim to retain normal tissue architecture with as little trauma as possible, necessitating careful dissection and specimen removal (23,24). Silicone oil may be used to tamponade the biopsy site following the procedure (24).
PVRL is rare enough that it does not yet have a standardized evidence-based treatment regimen (5,13). Clinical treatment trials often contain only handfuls of patients and include multi-pronged treatment regimens, making it difficult to determine optimal therapy to improve survival and minimize morbidity. General treatment considerations include whether there is unilateral or bilateral ocular involvement, presence of concurrent CNS disease or systemic disease, prior therapy and, importantly, patient goals. Treatment regimens are often individualized and involve collaboration between ophthalmologists, neuro-oncologists, and pathologists for these reasons (2,5).
PVRL has demonstrated sensitivity to both radiation and chemotherapy, and achievement of partial or complete remission is possible in many cases (2,13,17). Therapy may be local in the form of intravitreal chemotherapy and/or ocular radiation, while systemic treatment with chemotherapy and/or whole brain radiation are also utilized. In the case of isolated PVRL without evidence of CNS or systemic lymphoma, there is controversy over whether systemic treatment should be initiated (1,5,30). The largest clinical research studies of 83 and 78 PVRL patients without evidence of brain involvement found no difference in relapse, progression-free survival or overall survival between patients treated with local therapy vs patients treated with systemic therapy. Addition of systemic therapy in this patient population may therefore result in greater toxicity without overt benefit (15,18,30,31). However, both studies were retrospective and the wide array of treatment regimens within the categories of local and systemic therapy limits generalizability. The high incidence (65–90%) of eventual or occult CNS disease which carries grim prognostic implications remains justification for systemic therapy (1,5,20,32). The European Association of Neuro-Oncology has deemed either local chemotherapy or systemic chemotherapy to be acceptable treatment regimens for PVRL without CNS disease, while the International PCNSL Collaborative Group for PVRL recommends local therapy for isolated ocular disease (15,33).
The backbone of treatment for PVRL with concurrent CNS lymphoma is high-dose systemic methotrexate-based chemotherapy (2,30,33). Additional systemic agents are often administered, such as rituximab and cytarabine, though again lack of randomized controlled treatment trials preclude the determination of optimal chemotherapeutic regimen (10,33,34). Local intravitreal chemotherapy and/or ocular radiation is often added as adjunct therapy to target intraocular disease (1,13). Intravitreal chemotherapy is most commonly methotrexate, given in three phases (induction, consolidation, and maintenance) with decreasing frequency between injections (35,36). Rituximab is increasingly utilized as chemotherapy, both systemically and intravitreally, as research suggests it may offer less toxicity in comparison to methotrexate (37-39). Adverse effects of intravitreal rituximab include transient intraocular pressure elevation and iridocyclitis (38,39). Intravitreal methotrexate has been associated with significant retinal and corneal toxicity. Keratopathy is a frequent adverse effect of intravitreal methotrexate which may become severe, manifesting as blurred vision, redness, photophobia and excessive tearing (30,35,36). Whole-brain radiation therapy (WBRT) is also utilized, though neurotoxicity is a major concern particularly in patients over 60. More recent research suggests that disease control with less morbidity may be achieved with chemotherapy alone and WBRT should be reserved for chemoresistant or relapsing disease (1,2).
Many experimental therapies and treatment modalities are under investigation. Intrathecal chemotherapy with methotrexate has been studied, yet additional clinical trials are needed to parse out potential benefit. Various chemotherapeutics such as lenalidomide, thiotepa, ibrutinib and others and autologous stem cell transplantation have been studied for relapsing disease (40-42).
PVRL is an aggressive malignancy. Five-year survival rates have been estimated to be less than 25% and overall survival from diagnosis has been estimated to be around 31 months or less, but in recent years the outlook for PVRL treatment may be improving (6,31). One recent study estimated 5-year survival at 41.4% for patients diagnosed with primary B-cell lymphoma of the eye and ocular adnexal regions (19). The somewhat higher survival rate may be attributable to evolving chemotherapy regimens over time, but further research will be necessary to determine this. Relapse and recurrence are frequent, and the most common cause of death for PVRL patients is ultimately PCNSL (18,30). Management of PVRL patients should therefore include MRI with contrast every three months to screen for development or progression of CNS disease (13).
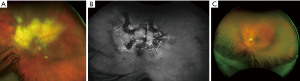
Primary uveal lymphomas are IOLs arising in the choroid, iris, or ciliary body. These are a separate entity from vitreoretinal lymphomas etiopathologically and have no association with CNS lymphoma. Primary choroidal lymphoma is by far the most common of these, yet all are extremely rare (9,43). Primary lymphoma of the ciliary body alone is almost unmentioned in current literature and will be discussed under the broader context of primary uveal lymphoma in this paper (4,9). Primary lymphomas affecting only the iris exist in a handful of case reports, after excluding the more frequent cases of iris lymphoma which are suspected to have arisen secondary to systemic or choroidal lymphoma (4,44). Cases of suspected primary iris lymphoma are often high-grade B-lymphocytic neoplasms which carry poor prognosis (4,9). In contrast, primary choroidal lymphomas are most often indolent low-grade B lymphocyte neoplasms histologically categorized as extranodal marginal zone lymphoma (EMZL). They were previously termed “uveal pseudotumors” or thought to be lymphoid hyperplasia due to their low-grade histology and low metastatic potential (1,7,16,43). Involvement of ocular adnexal structures can frequently be seen however as these tumors tend to infiltrate transsclerally to cause epibulbar extensions. These can present as crescentic thickening or as discrete masses (5,7,8,43). Cases of primary choroidal lymphoma appear to show a predilection for males, ages 50–70, who present with blurred vision or metamorphopsia. Occasionally patients may be asymptomatic, and disease is detected on ophthalmologic exam (7-9,20). Primary iris lymphoma is often misdiagnosed as anterior uveitis, given the overlapping symptoms of blurred vision, redness and eye pain (4,9,44).
Funduscopic examination shows evidence of multiple pink-yellow or creamy yellow choroidal swellings when lymphoma involves the choroid (Figure 5A-5C). Lymphoma cells may frequently infiltrate through the sclera to cause subconjunctival ‘salmon-patch’ lesions (9,44,45). When the iris is involved (Figure 5D), there may be ill-defined iris stromal thickening. Involvement of the iris and ciliary body is associated with more aggressive disease and is more commonly seen in secondary metastatic uveal lymphoma rather than primary (3,8). Features indicative of iris lymphoma rather than anterior uveitis include hyphema, abnormal iris vessels, and the aforementioned salmon-patch conjunctival lesions (9,44). The uveal tract may become diffusely thickened and swollen with disease progression. Choroidal folds and placoid infiltrates may be present (9,43). Fluorescein angiography and indocyanine green angiography (IGA) (46) often show hypofluorescent lesions; a study of 22 patients found hypofluorescent lesions on IGA corresponding to the observed choroidal lesions in 100% of cases (43). Additional findings which may be seen include hyphema, pseudohypopyon, anterior chamber reaction, and keratic precipitates. Infiltration of the iris and angle structures may lead to secondary glaucoma if untreated (1,3,45).

The definitive diagnosis of primary uveal lymphoma again relies on histopathologic confirmation of malignant cells (4,43). Biopsy of epibulbar, conjunctival or anterior chamber lesions is less invasive than posterior segment biopsy, and can provide convenient, easily accessible samples for diagnosis, but these may be lymphoproliferative or reactive inflammatory cells. More invasive biopsy of choroidal lesions may be necessary to obtain cytopathologic diagnosis (8). Flow cytometry, PCR and immunohistochemical staining for B-cell markers are useful to confirm cellular lineage and monoclonality (1,3,45).
Diagnosis of primary uveal lymphoma may reach a high degree of clinical suspicion based on characteristic funduscopic and imaging findings. B-scan ultrasonography is the most useful imaging tool in evaluation for uveal lymphoma, which can visualize hollow choroidal lesions and extrascleral extension which may otherwise be undetected. The combination of hollow choroidal thickening and acoustically hollow epibulbar extensions is highly suspicious for choroidal lymphoma (3,8,43). Uniform enhancement on computed tomography (CT) or MRI is typical in cases which have extrasceral extension. However, these imaging modalities are less useful due to the ill-defined appearance (47). Angiography also has demonstrated diagnostic benefit, in particular IGA for visualization of abnormalities in the choroid circulation (43). Anterior segment OCT and ultrasound biomicroscopy are useful tools to differentiate lymphoma of the iris from anterior uveitis, though biopsy is ultimately necessary for definitive diagnosis (4). All patients should undergo complete evaluation for systemic lymphoma at diagnosis including complete blood count, metabolic panel, CT chest/abdomen/pelvis, and sometimes bone marrow biopsy or positron emission tomography (PET) imaging (8,43,44).
Management of primary uveal lymphoma is nonstandardized given its rarity and most often involves discussion via multidisciplinary tumor board (4,43,45). Treatment with external beam radiotherapy (EBRT) may induce complete, lasting remission for primary choroidal lymphoma and is well tolerated (45). Prognosis is generally excellent for primary choroidal lymphomas which are histologically EMZL, with small studies finding 5-year survival rates to be 100% and rates of complete remission up to 79% in cases managed with either radiotherapy or rituximab (8,43,45). Given the lack of literature specific to primary iris and/or ciliary body lymphoma and the lack of certainty that existing cases are truly primary lymphomas, it is difficult to recommend optimal management or predict prognosis (9,43,44). Observation can be an acceptable management option in elderly patients with slowly progressive disease (4). However, untreated primary uveal lymphoma can be locally invasive and lead to serious complications such as glaucoma and vision loss and necessitate enucleation; therefore close follow up is recommended (3). Though dissemination is extremely rare, it is recommended for patients to undergo annual evaluation for relapse and/or systemic lymphoma (8,44).
The final category of IOLs comprises those which arise outside the CNS and metastasize to intraocular structures to cause secondary IOL. The choroid is the most common site to be affected by secondary IOL, likely through hematogenous spread through choroidal circulation (15). Secondary IOL is often confined to the choroid alone, and it is hypothesized that Bruch’s membrane may serve to contain the lymphoma cells. The iris and ciliary body are involved more rarely (44). Metastasis of systemic lymphoma to vitreoretinal structures is extremely rare (5,9,15).
Like the majority of systemic lymphomas in general, the majority of lymphomas which metastasize intraocularly are of B-cell origin. The diffuse large B-cell lymphoma (DLBCL) histologic subtype is the most common secondary IOL (9). It is generally a high-grade aggressive neoplasm, and it is not uncommon for additional systemic and CNS metastases to be present if there has been intraocular metastasis (5,8,10). Secondary IOL of T-lymphocyte origin is rarer but does occur, often from metastasized cutaneous T-cell lymphomas such as mycosis fungoides (48).
Metastatic IOLs usually appear as pale choroidal lesions with associated subretinal fluid. There can be mottled brown spots which may look similar to the “leopard spotted” appearance of PVRL. Anterior chamber cells and keratic precipitates are common when there is involvement of the iris (44). Vitreoretinal structures are usually not involved (16).
Diagnosis of secondary IOL can be suspected in patients with a known history of systemic lymphoma presenting with new ophthalmic symptoms, but is definitively achieved through histopathologic examination of the involved intraocular tissues (48). Management is dependent upon the underlying primary malignancy and generally will involve systemic therapy, with adjunctive intravitreal chemotherapy in some cases (49). Due to the potential for extensive disease at diagnosis, enucleation may be a necessary component of management (50). Secondary IOL often represents advanced metastatic disease by nature. Prognosis for secondary IOL will rely upon the primary malignancy but is typically poor (9,50).
IOL represents a group of multiple malignancies with distinct clinicopathologic features (Table 2). Vitreoretinal lymphomas are generally primary malignancies, originated from B-lymphocytes, associated with CNS lymphoma, characterized by an aggressive clinical course, and are treated with evolving methotrexate-based therapies. Primary uveal lymphomas are most commonly choroidal in location, associated with extrascleral extension and ocular adnexal involvement, originate from low-grade B-lymphocytes, and are treated with EBRT with good prognosis. Secondary IOLs typically metastasize to the choroid and carry a poor outcome due to associated widespread disease. Diagnosis of all types of IOLs is based upon cytologic detection of malignant lymphocytes and aided with immunohistochemical staining, PCR and flow cytometry. Future outlook for treatment and prognosis of IOL is likely to improve with less invasive molecular diagnostic techniques and increased awareness. Clinicians should be circumspect in all patients with possible IOL and promptly refer to oncologic specialists for rapid evaluation and treatment.
| Variable | Primary vitreoretinal lymphoma | Primary uveal lymphoma | Secondary intraocular lymphoma | ||
|---|---|---|---|---|---|
| Primary choroidal lymphoma | Primary iris lymphoma | Primary ciliary body lymphoma | |||
| Epidemiology | M = F, 6th decade of life, particularly immunodeficient and immunosuppressed | M > F, ages 50–70 | Unknown | Patients with known systemic lymphoma | |
| Malignant cell histology | Large B lymphocytes, rarely T lymphocytes | Low-grade B lymphocytes | High-grade B lymphocytes | N/A | Large B lymphocytes, rarely T lymphocytes |
| Associations | Central nervous system lymphoma | Not associated with CNS lymphoma | Systemic lymphoma, rarely cutaneous T-cell lymphoma (mycosis fungoides) | ||
| Ophthalmic findings | Vitreous haze, vitreous cells, retinal pigment epithelium changes with subretinal deposits (“leopard spotting”), often without cystoid macular edema | Multiple pink-yellow to creamy yellow choroidal swellings, subconjunctival salmon-patch lesions, diffuse thickening of the uveal tract, hyphema, abnormal iris vessels, choroidal folds, anterior chamber reaction, keratic precipitates | Typically confined to the choroid; pale choroidal lesions with associated subretinal fluid | ||
| Diagnostics | Tissue biopsy (pars plana vitrectomy, chorioretinal biopsy); adjunctive molecular diagnostic tools such as PCR, IHC, microRNA expression, cytokine analysis of aqueous or vitreous fluid interleukins; MRI and CSF analysis for detection of concurrent CNS disease | Tissue biopsy (choroidal lesions, or epibulbar/conjunctival/anterior chamber lesions if present); adjunctive B-scan ultrasonography, IGA, OCT, ultrasound biomicroscopy | Tissue biopsy; diagnosis can be presumed in patients with known systemic lymphoma and new ophthalmic symptoms | ||
| Management | May include local intravitreal chemotherapy and/or ocular irradiation as well as systemic chemotherapy and/or whole brain radiation; chemotherapy is often methotrexate-based with rituximab becoming more frequently utilized | Multidisciplinary and individualized; may include radiotherapy, chemotherapy and/or observation | Dependent upon primary malignancy | ||
CNS, central nervous system; PCR, polymerase chain reaction; IHC, immunohistochemistry; MRI, magnetic resonance imaging; CSF, cerebrospinal fluid; IGA, indocyanine green angiography; OCT, optical coherence tomography.