Background: The ex vivo model represented by mouse retinal explants in culture is a useful experimental model to investigate the molecular mechanism involved in neurovascular diseases such as diabetic retinopathy (DR). It ensures an experimental overview with more complete respect to isolate cells and reduce problems in terms of accessibility and management with respect to in vivo model. In particular, it allows the evaluation of the relationship between retinal cells in response to the typical stressors involved in DR pathogenesis.
Methods: Ex vivo retinal fragments derived from 3- to 5-week-old C57BL/6J mice. In particular, after dissection, the retina is cut into 4 separate fragments and transferred onto inserts placed with ganglion cells up. Once in culture, the explants could be treated in stress conditions typical of DR. In particular, this study protocol describes the procedure for the preparation and the culture of retinal explants with specific metabolic stressors such as high glucose (HG), advanced glycation end product (AGE), and oxidative stress (OS). In the end, this paper provides the protocols to perform molecular analyses in order to evaluate the response of retinal explants to stress and/or neuroprotective treatments.
Discussion: The cultured retinal explants represent an ex vivo experimental model to investigate the molecular mechanisms involved in neurovascular diseases such as DR. Moreover, they could be useful to test the effect of neuroprotective compounds in response to metabolic stressors in a fewer time respect to an in vivo model. In conclusion, retinal explants in culture represent a valuable experimental model to conduct further studies to better understand the pathophysiology of DR.
The ex vivo experimental model consisting of mouse retinal explants in culture could be defined as a halfway between an in vitro and an in vivo model (1). It allows to study the relationships between different retinal cells in a controlled environment. This model ensures the strict control of physicochemical parameters typical of in vitro cell cultures while maintaining the complexity, the integrity, and the main physiological properties of the whole organ. Retinal explants in culture allow to study the effects induced by metabolic stressors such as high glucose (HG), advanced glycation end products (AGEs) and oxidative stress (OS), which are involved in the pathophysiology of neurovascular retinal diseases like diabetic retinopathy (DR) (2,3). In particular, in DR the high glycolytic flux induces the increase of intermediate metabolites that could activate harmful pathways, such as the AGE pathway. The bind of AGEs with their receptor RAGE leads to inflammation, vascular dysfunction, glial activation, and neuronal apoptosis (4,5). In addition to the AGE pathway, in DR other intracellular cascades are activated resulting in OS condition (Figure 1).
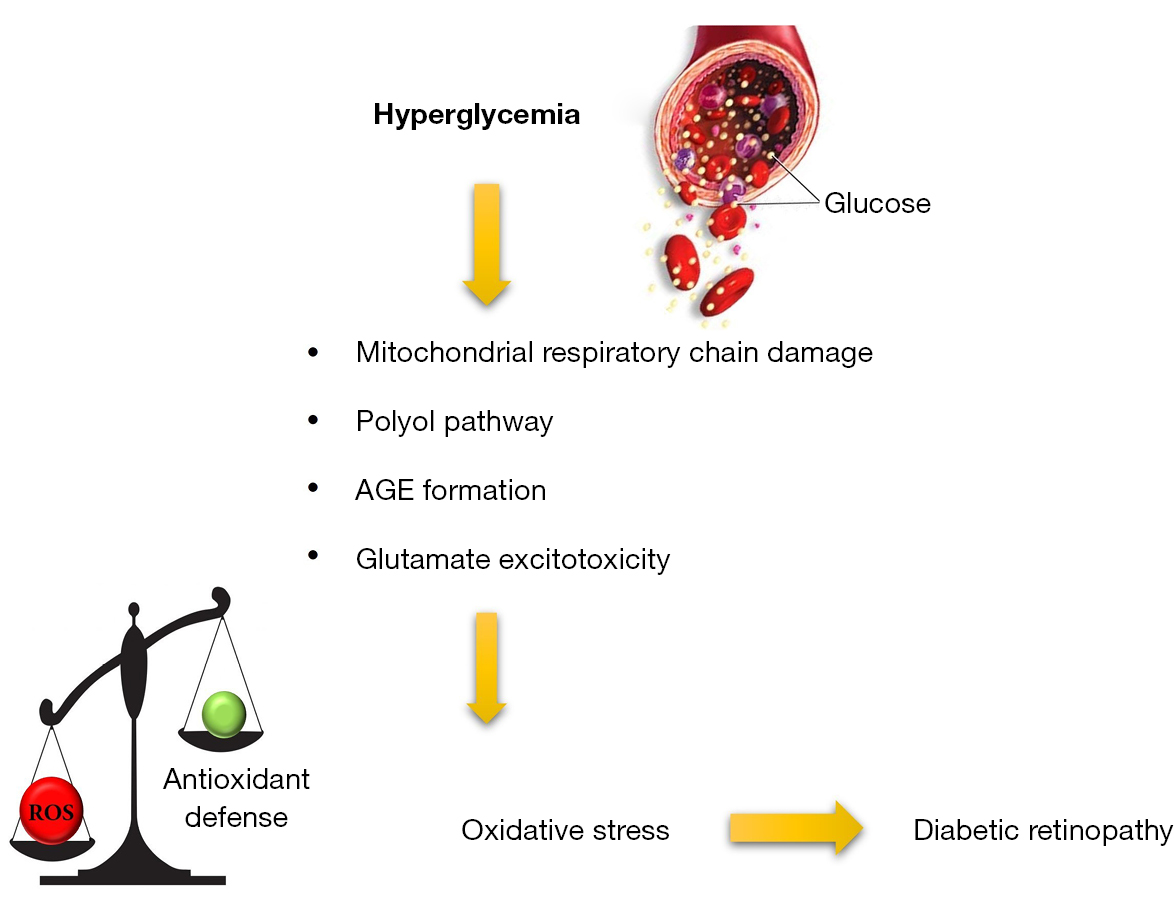
OS is a common etiopathogenic trigger involved not only in DR but also in other retinal diseases (6). OS is due to an imbalance between reactive oxygen species (ROS) production and antioxidant defence systems such as antioxidant phase II enzymes whose expression is regulated by nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2), the master regulator of the antioxidant response (7,8). OS-induced metabolic changes lead to alterations in function and structure of retinal neurons and microvasculature (9,10). Although the molecular mechanisms involved in neuronal, glial, and vascular changes caused by metabolic stressor in retinal diseases have been widely studied, there are still several points that remain elusive and need to be clarified. For these reasons, ex vivo models could be used to investigate specific molecular mechanisms involved in neurovascular diseases such as DR. Indeed, these models allow to define both the concentration of the stressors and/or the exposure time modulating the final entity of the stress (2). Similarly, and concomitant to stressors, also drugs can be dissolved in the culture medium to debug and test possible therapeutic strategies to counteract the onset of neurovascular diseases (11,12).
The protocol described refers to an ex vivo model tuned and validated by Amato et al. (2) 2016. In the same way, the real-time PCR, Western blot, immunofluorescence and reactive oxygen species (ROS) detection described protocols have been used to obtain the results published in the papers cited below.
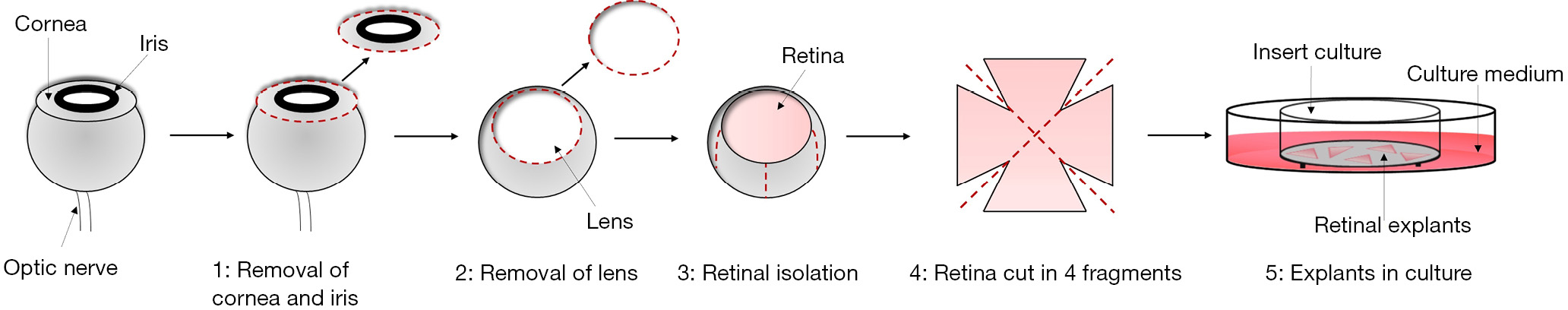
Ex vivo retinal explants are prepared using 3- to 5-week-old C57BL/6J mice (Charles River Laboratories, Wilmington, MA, USA) maintained in a regulated environment (23 ℃, 50% humidity) with a 12-hour light/dark cycle. After sacrifice through cervical dislocation, the eyes are enucleated using specific Castroviejo Spring Scissors—Sharply Curved Up (No. 15017-10, Fine Science Tool, Heidelberg, Germany). Once enucleated, eyes are collected in a 35 mm diameter sterile Petri dish filled with phosphate buffered saline (PBS 1X). The retinal dissection is performed in minimum essential medium (MEM; Merk/Sigma-Aldrich, Darmstadt, Germany) using a stereoscope (Nikon, Europe, Amsterdam, The Netherlands). The timeframe between mouse scarification and retinal preparation is about 10–15 min.
To perform retinal dissection, the following instruments may be used:
Retinal dissection consists of different subsequent steps:
After retinal dissection, any residues of corpus vitreous and/or trabecular meshwork are removed, and the retina is cut into 4 separate fragments using straight scissor. The fragments are then transferred onto Millicell-CM culture inserts (Merck Millipore, Darmstadt, Germany; Figure 2) with ganglion cells up, which means that the ganglion cell layer is facing up and the photoreceptors are in contact with the filter of the culture insert. The inserts are placed in six well tissue culture plates containing 1 mL of culture medium composed by:
Culture medium and antibiotics are from Merck/Sigma Aldrich.
The explants are incubated at 37 ℃ under a humidified 95%/5% (vol/vol) mixture of air and CO2.
Usually, the number of retinal explants transferred onto each insert depends on the molecular analysis that is planned for that experiment. In particular, for real time PCR or western blotting assay, 8–10 explants are needed to be pulled and ensure the extraction and purification of an adequate amount of RNA or proteins. Conversely, for immunohistochemistry, 4 retinal explants are sufficient to obtain an adequate number of retinal sections.
As described by Amato et al. (2), the ex vivo model allows to study the effect of metabolic stress conditions such as HG, OS, or AGEs.
The HG condition is tuned adding 75 mM D glucose (Merk/Sigma) in the culture medium and changing the medium every other day; 75 mM been demonstrated to be the effective glucose concentration able to induce hyperglycaemic retinal damage. In particular, the apoptotic rate increased after 5 days reaching the maximum levels after 10 days of incubation (2).
The AGE treatment is performed adding 100 μg/mL AGE-BSA (BioVision, Milpitas, CA, USA) to the culture medium and changing the medium every other day. In this case, the apoptotic rate has been shown to be significantly higher than in control explants after 7 days of incubation (2).
OS can be induced adding 100 μM H2O2 (Merk/Sigma) in the culture medium and changing the medium every day. In this condition, a significant increase of apoptosis in the retinal explants has been observed after 5 days of incubation (2).
Considering the crucial role of hyperglycaemia in the pathophysiology of DR, the HG condition could be the most appropriate treatment to study the molecular alterations induced by diabetes. On the other hand, the OS and AGE treatments also reproduce conditions that are closely related to hyperglycaemia and DR. Indeed, hyperglycaemia is the main causative effect of OS and AGE accumulation, which, in turn, are involved in retinal neuronal death and glial dysfunction. Therefore, culturing retinal explants in HG conditions allows a more complete representation of the pathophysiological events involved in DR progression, but this requires a relatively long culture time (10 days). Conversely, AGE and OS treatments need shorter culture times than HG treatment and allow to investigate the specific role of these metabolic stressors in the pathogenesis of DR. In other words, the settings and the type of treatments are strongly correlated with the experimental plan. For example, if the aim is to test the antioxidant properties of a specific compound, the most suitable experimental condition is represented by OS treatment.
To evaluate the effect of metabolic stressor on neuronal death, inflammation, and OS in retinal explants, different molecular analyses may be performed. Here, I report the protocols used for real time PCR, western blotting, and immunofluorescence.
The explants on each insert are collected and stored at ?80 ℃. Total RNA isolation usually is performed with Rneasy Mini Kit (Qiagen, Hilden, Germany). Briefly, chemical lysis of the cell membranes is performed in 350 μL of RLT lysis buffer while the mechanical tissue lysis is performed with 3×10 s sonication cycles at 60 Hz. Following tissue homogenization, the RNA is purified using RPE and RW1 wash buffer. Subsequently the RNA is resuspended in 30 μL of RNAse-free water. RNA amount and purity are evaluated through spectrophotometric measurement using Nanodrop? spectrophotometer (NanoDrop products, Wilmington, CA, USA). Subsequently, 1 μg of RNA is retrotranscripted in first-strand cDNA using QuantiTect reverse transcription kit (Qiagen). The RT-qPCR is performed with 10 ng of cDNA using SYBR green master mix (Bio-Rad Laboratories, Hercules, CA, USA).
The explants on each insert are collected and stored at ?80 ℃. Total protein extraction is obtained using RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktails (Santa Cruz, Dallas, TX, USA). The chemical cell membrane lysis is recommended in 100 μL of RIPA buffer while the mechanical tissue lysis is performed with 3× 10 s sonication cycles at 60 Hz.
The extraction of separate cytoplasmic and nuclear protein fractions ca be performed using a NE-PER? Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). The extraction of the cytoplasmatic fraction is recommended in 200 μL of Cytoplasmic Extraction Reagent I and the tissue homogenization can be made using Dounce homogenizer or a tissue grinder. Alternatively, a syringe with a 26- or 27-gauge needle can be used.
Once purified, the amount of isolated protein is quantified using a MicroBCA assay kit (Thermo Fisher Scientific).
Western blotting is performed using 30 μg of total protein, 10 μg of cytosolic protein fraction, or 5 μg of nuclear protein fraction.
Mouse retinal fragments are fixed in 4% paraformaldehyde (Merk/Sigma) in 0.1 M phosphate buffer for 2 h at room temperature, and then they are stored in 25% sucrose (Pan Reac Appli Chem, Darmstadt, Germany) in 0.1 M phosphate buffer at 4 ℃. To proceed with cryostat sections, explants are positioned in specific molds and are embedded in cryo-gel (Bio Optica, Milan, Italy). Then they are frozen in liquid nitrogen and stored at ?20 ℃. Subsequently, retinal fragments are cut into 10-μm-thick coronal sections with a cryostat and the sections are mounted onto gelatinized slides. Alternatively, can be used SuperFrost microscope slides (Thermo Fisher Scientific)
The sections are incubated overnight at 4 ℃ with primary antibodies and then with secondary antibodies (usually at a dilution of 1:200) at room temperature for 2 h. Primary and secondary antibody are diluted in PBS containing 0.5% Triton X-100. Between primary and secondary antibody incubation, slides are washed in PBS (3× 5 min). To label the nuclei of retinal cells, the slides can be coverslipped with Fluoroshield Mounting Medium containing DAPI (Abcam, Cambridge, UK).
Detection of intracellular ROS can be performed using the 2′,7′-dichlorofluorescein diacetate (DCFH-DA) assay. DCFH-DA is a hydrophobic non-fluorescent molecule that freely enters the cells and is rapidly hydrolysed to DCFH by an intracellular esterase. In the presence of ROS, DCFH is oxidized to its fluorescent product (DCF) (13). DCF fluorescence can be visualised with a fluorescence microscope and its intensity can be evaluated with fluorimetric methods.
The following protocol may be used to detect intracellular ROS in OS-treated retinal explants:
Taken together, the techniques described here are useful to investigate the molecular mechanisms triggered in response to metabolic stress typical of DR. For instance, the ex vivo retinal explant models may allow to clarify different points regarding:
In detail, in retinal explants treated with HG, AGEs or OS a significant increase of apoptotic activity was demonstrated with RT-qPCR measurements of increased caspase-3 mRNA levels, which correlated with higher cytochrome c and cleaved caspase-3 protein levels evaluated with western blotting (2,12,14). In addition, in HG treated explants apoptotic cell death was found to be associated to a significant decrease of autophagy and upregulation of mTOR (14). Moreover, active caspase-3 immunolabeling in HG-, AGEs- and OS-treated explants suggested that in retinal explants apoptotic neurons are mainly localized in the inner nuclear layer and ganglion cell layer. In addition, an analysis of DAPI-labeled retinal layers demonstrated that the exposure to metabolic stresses does not cause gross morphological changes in the layer thickness of the retinal explants (2,14). Concerning the OS-treated explants, as demonstrated by DCF assay, we recorded an increase of intracellular ROS levels. This correlated with an increase of antioxidant enzyme mRNA levels, while their protein amount was significantly reduced, suggesting that retinal explants in OS undergo an intense turnover of antioxidant enzymes driven by Nrf2. Indeed, in OS condition, retinal explants undergo to an increase in nuclear protein levels of Nrf2 that is associated with its mRNA upregulation (11). We also evaluated the effect of metabolic stressors on VEGF, a key growth factor in the pathogenesis of DR. Using the ex vivo model of retinal explants, we demonstrated that VEGF plays a neuroprotective role during the early phases of DR. Nevertheless, if the metabolic stress persists, VEGF promotes an increase of its own expression and release through an autocrine loop (3). Taken together, these results suggested that both the dose of metabolic stressor and the time of culture are adequate to induce in retinal explants the typical condition of an early phase of DR. In the last few years, we tested the therapeutic efficacy of neuropeptides such as octreotide, a somatostatin analog, and natural compounds such as Lisosan G, a fermented powder obtained from whole grains (2,12,14). In both cases, the studies performed on ex vivo models of retinal explants were useful to understand the neuroprotective and/or antioxidant potential of these compounds. In particular, studies in OS treated retinal explants to evaluate the protective effects of Lisosan G were useful to tune the effective dose of natural compound able to prevents neuronal death and OS. These results were pioneers for in vivo studies using diabetic rats (12). Indeed, in this case we found a strong correlation and consistent between ex vivo and in vivo studies.
In conclusion, cultured retinal explants represent a reproducible and useful ex vivo experimental model to investigate the cell-cell interaction, the retinal structural organization, and the specific molecular mechanisms and pathways involved in the onset of neurovascular diseases. It is a joining link between in vitro and in vivo models. The use of organotypic retinal culture combines the advantages of using cell cultures and animal models. Moreover, it allows a more complete experimental design with respect to isolated cells and greater accessibility and management with respect to an in vivo model. Retinal explants in culture are essential in fine-tuning and testing the effect of specific stressors and pharmacological compounds in relatively short time compared to the periods required to ensure the onset of a chronic disease such as DR in an in vivo model. On the other hand, the ex vivo experimental model simplifies the pathophysiological conditions and due to the limited life span of the isolated tissue, can be maintained in culture for a limited time. Moreover, it does not consider signalling from the surrounding environment and bloodstream. For this reason, in order to confirm and better clarify the results obtained with retinal explants, it is also necessary to use microfluidic approaches and/or in vivo models of DR such as streptozotocin-induced diabetic mice, Akita mice or db/db mice.