Abstract: Autoimmune retinopathy (AIR) refers to both paraneoplastic and non-paraneoplastic forms of a rare, acquired retinal degeneration thought to be mediated by the production of antiretinal antibodies. However, the mechanisms underlying AIR pathogenesis are incompletely understood, and it remains a diagnosis of exclusion given the lack of definitive testing as well as its protean clinical presentation. This review summarizes the current literature on the epidemiology, diagnosis, and management of AIR, with a focus on non-paraneoplastic disease and the potential role of immunomodulatory therapy. A recent expert consensus statement on diagnosis and management of non-paraneoplastic AIR served as a framework for interpreting the limited data available, a process that was complicated by the small sample sizes, heterogeneity, and retrospective nature of these studies. Additional work is needed to characterize AIR patients on the basis of cytokine and immunogenetic profiling; to establish the pathogenicity of antiretinal antibodies; and to standardize treatment regimens as well as assessment of clinical outcomes.
Autoimmune retinopathy (AIR) refers to an acquired retinal degeneration, initially described in the setting of cancer and thought to be mediated by anti-recoverin antibodies (cancer-associated retinopathy and melanoma-associated retinopathy; collectively referred to as paraneoplastic autoimmune retinopathy, pAIR) (1,2). Subsequently, AIR has been reported in the absence of malignancy, in the context of other autoimmune diseases, and associated with other anti-retinal, anti-optic nerve, and anti-glycolytic enzymes (non-paraneoplastic autoimmune retinopathy, npAIR) (3). Given attention would be primarily directed towards treatment of the underlying malignancy in pAIR, this chapter focuses on the diagnosis and management of npAIR.
AIR is an exceedingly rare entity, and thought to account for <1% of all cases seen at tertiary referral centers (4). The protean nature of the disease and the lack of standardized diagnostic criteria until recently may have further contributed to both underdiagnosis and misclassification. To date, there are no studies on the prevalence of pAIR or npAIR. Understanding of the natural history, evaluation, and management of npAIR largely derives from the pAIR literature, case reports, and expert consensus (5,6).
Including patients who were later found to be seronegative for antiretinal antibodies, Adamus and colleagues showed that patients clinically diagnosed with AIR (57.5%, 111/193) were more likely to be women. In addition, clinically diagnosed npAIR patients (mean age 55.9 years) were younger on average compared to pAIR patients (mean age 62.0 years) (7). Ferreyra and colleagues reported on 24 consecutive npAIR patients who were all seropositive for antiretinal antibodies. The median age was 47 years (range, 11–78 years), 62.5% (15/24) were female, and 66.7% (16/24) had a personal history of autoimmune disease (6). Similarly, an unpublished retrospective case series of 24 seropositive npAIR patients at the National Eye Institute found that the median age was 51.5 years (range, 37–88 years), 79.2% (19/24) were women, and 45.8% (11/24) had a personal history of autoimmune disease. Thus, npAIR patients may be younger on average than pAIR patients. There may also be both a female predilection for and an autoimmune predisposition to npAIR.
Patients with npAIR typically present with painless, subacute vision loss out of proportion to measured visual acuity, characterized by symptoms of diffuse photoreceptor dysfunction including photopsias, scotomas, nyctalopia/photoaversion, dyschromatopsia, and metamorphopsia (8,9). Disease manifestation is usually asymmetrically bilateral, though several cases of unilateral AIR and presumed npAIR (clinical diagnosis without antibody testing) have been described (10-12).
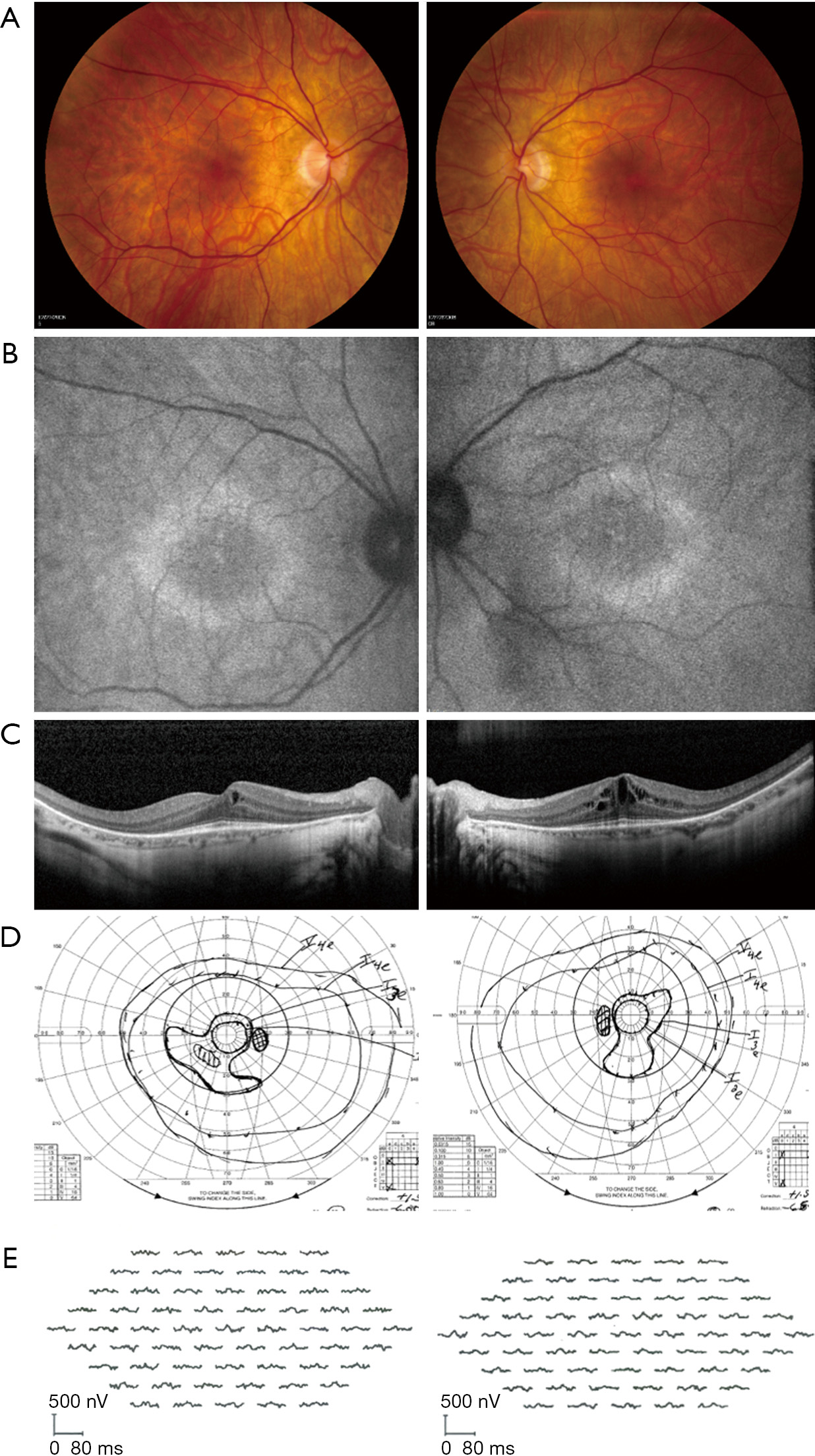
Depending on disease severity and duration, examination could be grossly unremarkable or could reveal signs of retinal degeneration including vascular attenuation, cystoid macular edema (CME), outer retinal/retinal pigmentary epithelium atrophy, and optic disc pallor. Bone spicule-like pigment deposits are uncommon. Electroretinogram (ERG) confirms abnormalities in scotopic/photopic responses, and may take on rod-cone, cone-rod, or electronegative configurations. Visual field testing shows corresponding scotomas and peripheral constriction (3,6,9).
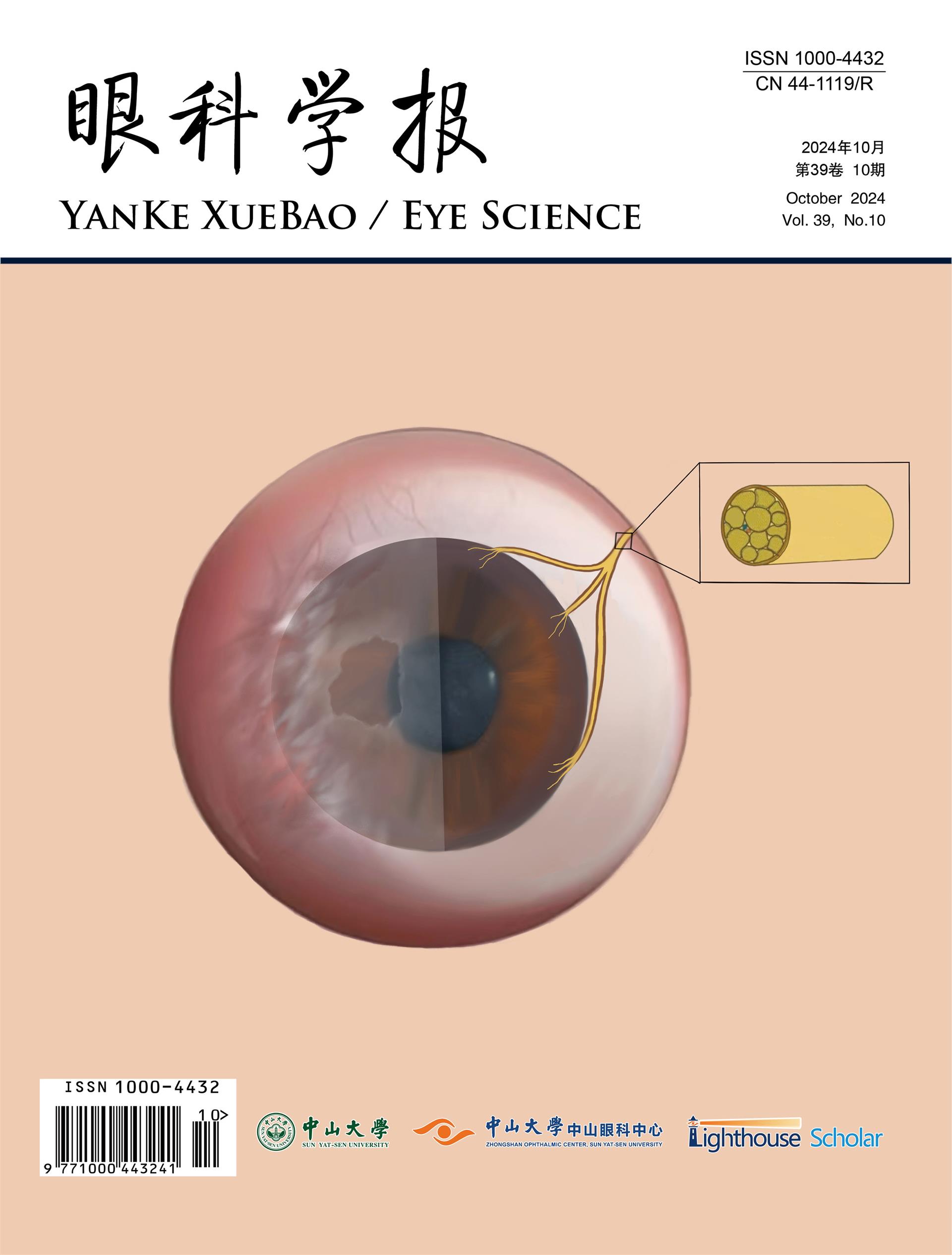
As seen in various retinal dystrophies, fundus autofluorescence may show a ring of parafoveal hyperautofluorescence, with associated outer nuclear layer thinning and ellipsoid zone attenuation on optical coherence tomography (OCT). There may also be patchy hypoautofluorescence in the periphery (Figures 1-3) (13-15). Fluorescein angiography is usually unremarkable (3). Mild retinal vascular leakage may be observed, but this is typically less than generally seen with primary retinal vasculitis, and there should otherwise be limited signs of intraocular inflammation (5,16).
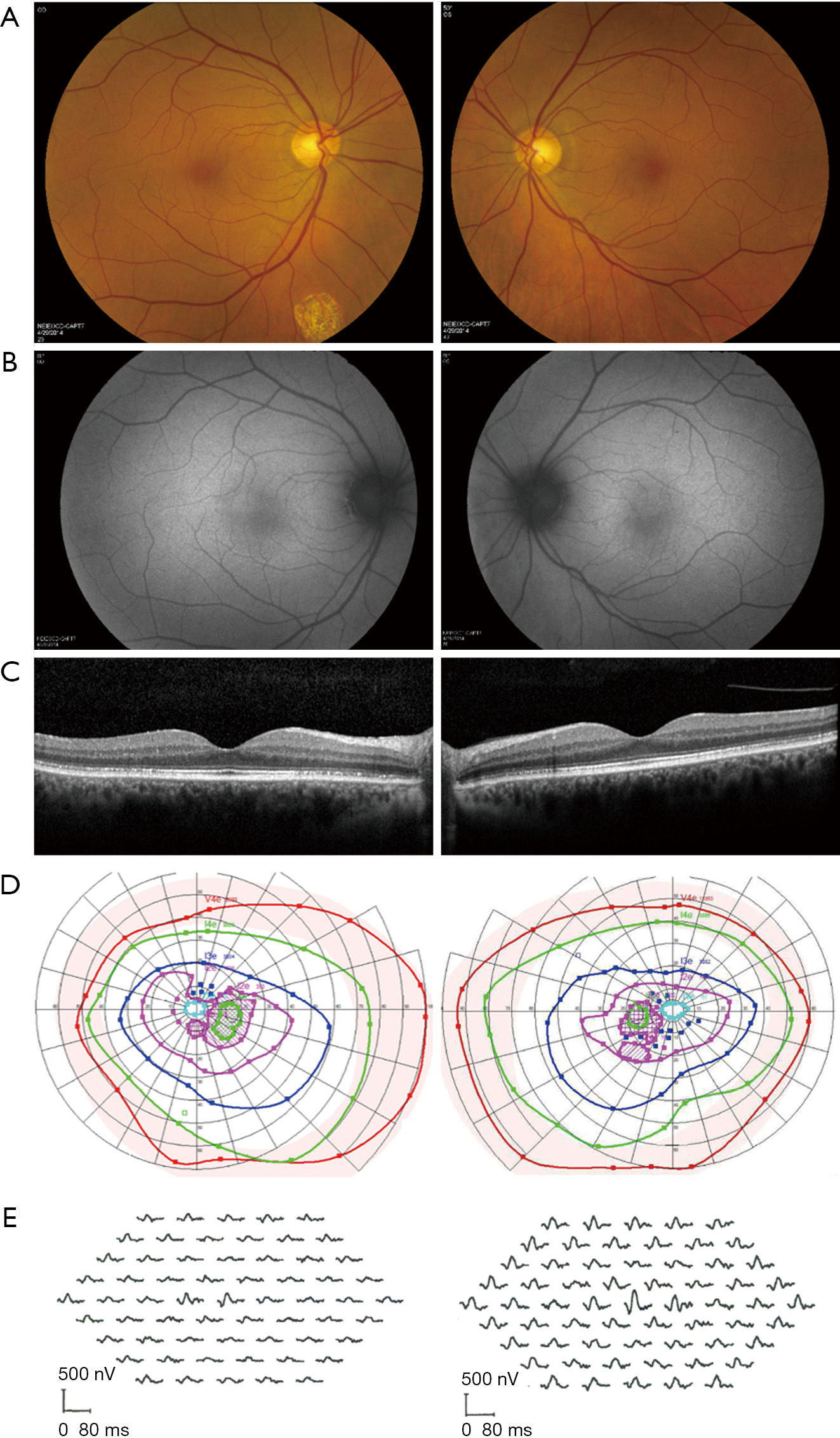
The prevalence of CME in npAIR is variable, with the two largest case series respectively reporting 45.8% (11/24) and 8.3% (2/24) (6). Prominent, recalcitrant CME despite use of multiple immunomodulatory agents has further been described (17,18). Ferreyra and colleagues suggested that CME could be a distinguishing factor in npAIR, such that patients with CME may have a subtype of disease that is more responsive to immunosuppressive therapy (6). Finn and colleagues assessed the presence of CME as a marker of npAIR progression using a retrospective series of 16 patients (32 eyes) with minimum follow-up of 1 year. Compared to eyes without CME at presentation and at final follow-up (n=21), eyes with CME at both time points (n=8) had lower maximal a-wave and b-wave amplitudes on ERG and faster rates of ellipsoid zone loss on foveal-centered spectral-domain OCT. Eyes with CME also exhibited worse visual acuity at presentation and at 1-year. Eyes with CME also had shorter ellipsoid zone length at presentation, which is not surprising (19). Interpretation of these results is complicated by the small/uneven sample sizes, as well as lack of adjustment for potential confounders such as age, disease duration, and differences in treatment administration/response. Additional research accounting for these factors is needed to clarify whether CME, ellipsoid zone length, and other imaging markers have utility as prognostic indicators in npAIR.
There is no definitive testing available for npAIR, and thus it remains a diagnosis of exclusion. Based on a consensus statement of 17 experts in 2016 (defined as ≥75% of the panel in agreement), essential diagnostic criteria for npAIR should include: (I) no history or examination findings indicative of another apparent cause of visual function abnormality); (II) ERG abnormality (with or without visual field abnormality); (III) presence of serum antiretinal antibodies; and (IV) absence of overt intraocular inflammation. Supportive diagnostic criteria include signs/symptoms of photoreceptor dysfunction, personal/family history of autoimmune disease, and rapid onset of vision changes. Notably, age at presentation and response to immunosuppressive therapy were not selected as supportive diagnostic criteria. In addition to obtaining a detailed history and thorough examination, all experts agreed that the initial evaluation of npAIR should therefore include ERG, fundus autofluorescence, serum antiretinal antibody testing (ideally via a two-tiered assay utilizing Western blot and immunohistochemistry), and comprehensive oncological workup/surveillance by an appropriate physician. Most experts also considered OCT and fluorescein angiography to be essential for diagnosis (Table 1) (5).
| Essential diagnostic criteria |
| No other apparent cause of visual dysfunction |
| Absence of fundus lesions/changes suggestive of alternative etiology, including hereditary retinal dystrophy |
| Absence of overt intraocular inflammation (<1+ anterior chamber or vitreous cells, <1+ vitreous haze) |
| Electroretinogram abnormality (with or without visual field abnormality) |
| Presence of serum antiretinal antibodies |
| Supportive diagnostic criteria |
| Signs/symptoms of photoreceptor dysfunction, including photopsias, scotomas, dyschromatopsia, nyctalopia, and photoaversion |
| Rapid onset of vision changes (acute/subacute, <6 months) |
| Personal or family history of autoimmune disease |
| Core diagnostic tests |
| Electroretinogram |
| Optical coherence tomography |
| Fundus autofluorescence |
| Fluorescein angiography |
| Two-tiered serum antiretinal antibody testing (Western blot and immunohistochemistry) |
| Oncological workup/surveillance |
While the detection of circulating antiretinal antibodies is considered the sine qua non of npAIR, Adamus and colleagues found that only 41.1% (58/141) of patients whose samples were tested upon clinical suspicion for npAIR were seropositive, albeit more than a decade before the expert consensus statement. In the same study, antiretinal antibodies were significantly more prevalent, but also not ubiquitous, among patients with clinical suspicion for pAIR (63.5%, 33/52, P=0.009). Overall, repeat testing on 9 seronegative patients with worsening symptoms 1–2 months later showed the same results, while the effect of immunosuppressive therapy on the number of antiretinal antibodies in seropositive patients was variable, highlighting the unreliability of antiretinal antibodies as diagnostic or prognostic indicators (8). Moreover, the pathogenicity of antiretinal antibodies likely depends on host factors, the retinal microenvironment, and is epitope-specific. In certain scenarios, they may further be the consequence rather than the cause of neuroretinal dysfunction (20). Accordingly, antiretinal antibodies may be found in healthy individuals as well as patients with competing diagnoses, including posterior uveitis and hereditary retinal dystrophies (21,22). Finally, there could be significant variability in the detection and quantification of antiretinal antibodies between laboratories, and there is currently only one center in the United States offering commercial testing using a clinical laboratory improvement amendments (CLIA)-certified protocol (23,24). Taken together, these results emphasize that the presence of antiretinal antibodies alone cannot be used to confirm the diagnosis of npAIR or guide management decisions, as well as the need for a better understanding of the basic mechanisms underlying npAIR in order to develop standardized and accurate laboratory assays (25).
Corticosteroids and conventional non-alkylating immunosuppressive drugs are considered first-line treatment for npAIR. However, most experts surveyed felt that biologics and intravenous immunoglobulin (IVIG) are also appropriate options at any stage of disease. Consensus on the role of plasmapheresis was not achieved (5).
The current body of evidence for immunomodulatory therapy in npAIR management is summarized below. When npAIR and pAIR patients were grouped together in the original analyses, key statistics were recalculated for only npAIR patients using the published data.
Ferreyra and colleagues reported the outcomes of 24 npAIR patients, most of whom were treated with a combination of local/systemic corticosteroids, cyclosporine, and azathioprine. Using the criteria of ≥2 lines of improvement in Snellen visual acuity or ≥25% improvement in Goldmann visual field, 63% of patients (15/24) showed response to immunosuppression. Specifically, 50% (12/24) of patients exhibited improvement in visual field, and 25% of patients (3/12) showed improvement in visual acuity; no patient demonstrated improvement in both categories. ERG was not repeated routinely. Among the 15 responders, 4 received only local/systemic steroids; 2 received steroids and cyclosporine; 2 received steroids and azathioprine; 6 received steroids, cyclosporine, and azathioprine, and 1 received steroids, cyclosporine, mycophenolate, and infliximab. Follow-up time of responders ranged from 3 to 89 months. Treatment patterns were similar among the 9 non-responders, though 3 patients also received IVIG, likely reflecting treatment escalation. In comparison, 100% (6/6) of pAIR patients showed improvement with immunosuppression (6). However, an accompanying editorial and re-analysis of the data by Jampol and Fishman suggested less favorable results; 81% of npAIR patients had no improvement or worsening of visual acuity in at least one eye, and 62% of npAIR patients had no change or worsening of visual field in at least one eye. Additionally, these reviewers commented that a learning effect could in part explain observed improvements in visual field, as well as that potential treatment responses may not be sustained. The original analyses also included two patients with a known family history of retinitis pigmentosa, both of whom were categorized as responders (26).
There are also individual case reports and smaller case series supporting the efficacy of cyclosporine, mycophenolate, and azathioprine (27-31), though the relatively low number of nonresponsive patients suggests probable publication bias (9,32,33). Of note, there are no reports of methotrexate being used successfully for the treatment of npAIR (34,35).
Rituximab is a chimeric monoclonal antibody against CD20, a molecule expressed on the surface of B-cells at all stages of differentiation. Rituximab is thought to act mainly via depletion of B-cells via apoptotic signaling as well as both complement- and antibody-mediated cytotoxicity (36). In addition, Rituximab targets a subset of CD3+CD20+ T-cells skewed towards production of proinflammatory cytokines (37).
Fox and colleagues described the first successful use of rituximab for npAIR associated with systemic lupus erythematosus. A 53-year-old woman had presented with 8-month history of worsening vision despite receiving cyclosporine 100 mg TID, azathioprine 100 mg BID, and topical prednisolone acetate 1%. Her visual function continued to worsen over 3 years despite increasing cyclosporine to 200 mg BID and azathioprine to 200 mg daily, while adding adalimumab 40 mg every 2 weeks. The patient was then given two doses of intravenous rituximab 1,000 mg separated by a 2-week interval, followed by yearly maintenance infusions. Patient exhibited improvement in visual acuity (20/200 to 20/125 OU) and ERG one month after the first cycle, and then remained stable for 5 years. Her visual field defects were unchanged (38).
Maleki and colleagues reported on 12 eyes of 6 patients with antiretinal antibodies who received rituximab as monotherapy (n=2), with cyclophosphamide (n=2), with bortezomib (n=1), or with both cyclophosphamide and bortezomib (n=1). The four patients receiving combination therapy had eyes with severe vision loss (≤20/200), nearly extinguished visual fields, or flat/nearly flat ERG amplitudes at baseline. Rituximab was administered as 8 cycles of 375 mg/m2 weekly, followed by 375 mg/m2 monthly (treatment duration 8–20 months). Treatment response was defined as stability/improvement of visual acuity (±1 line on the Snellen chart), visual field (same mean and pattern deviation based on Humphrey Visual Field probability plots), and ERG (±25% of baseline). Using these criteria, no eye showed improvement in visual acuity, though visual acuity stabilized in 8/12 eyes (66.7%). Similarly, visual field and ERG stabilized/improved in 8/12 eyes, though the outcomes were not necessarily correlated (i.e., an eye could have worsening visual acuity and ERG, but a stable visual field). Though the authors suggested a possible negative trend in the average number of antiretinal and anti-optic nerve antibody bands, the number of patients and the differences were too small for a meaningful assessment. Furthermore, these analyses included two patients previously diagnosed with birdshot chorioretinopathy and one patient previously diagnosed with HLA-B27-positive panuveitis. Excluding these 6 eyes, the purported treatment response would be 3/6 (50.0%) for visual acuity, 4/6 (66.7%) for visual field, and 2/6 (33.3%) for ERG (39).
Boudreault and colleagues summarized the results of 5 presumed npAIR patients treated with rituximab, 2 at a dosage of 375 mg/m2 and 3 at a higher dosage (1,000 mg flat dose). Primary outcomes included change in visual acuity and ERG amplitudes, both assessed as the simple ratio of post-treatment to pre-treatment measurements (i.e., 1 = stability, >1 = improvement, and <1 = exacerbation). Of the three patients who exhibited stability/improvement on rituximab, a 61-year-old woman had previously failed treatment with mycophenolate mofetil and infliximab; a 10-year-old girl had transient response to IVIG, prednisone, mycophenolate mofetil, and cyclosporine; and a 70-year-old woman had worsened on mycophenolate mofetil and prednisone. One of the two non-responders was a treatment-na?ve 16-year-old female who did not improve with rituximab monotherapy (2 monthly doses of rituximab 1,000 mg). However, whole-exome sequencing revealed known pathogenic mutations in MSFD8, a gene that has been associated with neuronal ceroid lipofuscinoses and retinal degeneration (40). The other non-responder was a 65-year-old man initially diagnosed with late-onset retinitis pigmentosa, who was treated with mycophenolate mofetil for 7 months followed by two cycles of rituximab (750 mg weekly for 4 weeks; same regimen repeated 6 months later). This patient showed continued deterioration on visual field and ERG testing, and while he reportedly eventually stabilized on plasmapheresis, his history could also be interpreted as the natural progression of a hereditary retinal dystrophy (41). No genetic testing results were reported.
Davoudi and colleagues reported on 9 npAIR and 7 pAIR patients treated with rituximab. Overall, the rate of visual acuity decline was significantly slower after initiation of rituximab therapy. All 9 npAIR patients showed stability (n=7) or improvement (n=2) in visual acuity. Among the latter, one was a 21-year-old woman previously treated with a prednisone taper and mycophenolate mofetil, who experienced improvement after addition of rituximab. The other was a 69-year-old woman who received a prednisone taper followed by rituximab monotherapy. All seven patients who had stable visual acuity were also treated with cyclophosphamide, and two of these individuals also received IVIG pre- and post-rituximab. In addition, no npAIR patient experienced improvements in visual field (one worsened) or ERG (all were stable). The median follow-up time among npAIR patients was 16 months (range, 8–34 months). In comparison, the study also included 7 pAIR patients, of whom 4 remained stable and 3 worsened on rituximab; the use of concurrent immunosuppressive medications was similar between the two groups (42).
Uludag and colleagues described a 50-year-old man who presented with a 5-month history of positive scotomas and dyschromatopsia; visual acuity was 20/20 and visual fields were full OU. The patient was treated with 4 cycles of rituximab 375 mg/m2/week, and experienced improvement in both ERG parameters and subjective visual complaints. The patient was then given 6 cycles of cyclophosphamide 1 g/m2/month, with further improvement on ERG and no new examination findings after completion of therapy (43).
The central role of TNF-α in complex signaling networks regulating cellular survival, inflammation, and autoimmunity has been well-described (44). Notably, tumor necrosis factor-alpha inhibitors (e.g., etanercept, infliximab, certolizumab, adalimumab, and golimumab) have not been used in pAIR, presumably because of concerns regarding possible increased risk of secondary malignancies and infections (45,46). Based on a search of PubMed, only infliximab and adalimumab have been used in the setting of npAIR.
Ferreyra and colleagues reported on two cases of npAIR with CME treated with infliximab. The first patient was a 44-year-old woman who failed to respond to a regimen consisting of prednisone, subtenon methylprednisolone, and infliximab after 9 months of treatment. The second patient was a 46-year-old woman who improved after receiving subtenon methylprednisolone, mycophenolate mofetil, cyclosporine, prednisone, and infliximab over the course of 28 months (6).
As previously summarized, Boudreault and colleagues described a 61-year-old woman who had worsened on mycophenolate mofetil and infliximab, but then responded and was stable on rituximab for 5 years (41). Similarly, Fox and colleagues detailed a 53-year-old woman who did not respond to adalimumab after first failing cyclosporine and azathioprine. She subsequently responded to rituximab and was stable over 5 years (38).
IL-6 is a proinflammatory cytokine that has been implicated in germinal center formation, terminal B-cell differentiation, and upregulation of IgG production (47). Based on a search of PubMed, only two biologic agents targeting the IL-6 pathway have been used for treatment of npAIR. Tocilizumab (humanized monoclonal antibody) and sarilumab (full human monoclonal antibody) are both designed to act against soluble as well as membrane-bound IL-6 receptors.
A 46-year-old woman with presumed npAIR treated with intravenous tocilizumab 8 mg/kg every 4 weeks demonstrated improvement in visual acuity, angiographic leakage, macular edema and outer retinal architecture. She had exhibited clinical progression over 3 years despite administration of topical/periocular corticosteroids as well as immunosuppression with mycophenolate mofetil and rituximab. The patient had near-complete resolution of the previously recalcitrant macular edema after 5 sessions, and was reportedly stable at 11 months with continued monthly infusions. Moreover, OCT showed partial reconstitution of the ellipsoid zone OU, with corresponding improvement in visual acuity from counting fingers to 20/200 OD, and from 20/200 to 20/40 OS (17).
Similarly, a 29-year-old otherwise healthy woman treated with subcutaneous sarilumab 200 mg every 2 weeks demonstrated improvement in visual acuity, angiographic leakage, macular edema, and ERG amplitudes after worsening on a regimen including topical/periocular corticosteroids, azathioprine, and adalimumab. The macular edema improved markedly after the first treatment, and remained completely resolved at 6 months with biweekly injections. Visual acuity improved from 20/70 to 20/32 OD, and stable at counting fingers OS (reportedly limited by optic neuropathy and outer retinal attenuation) (18).
Bortezomib is a reversible dipeptide boronate proteasome inhibitor that induces Bcl-2 phosphorylation and cleavage, which in turn leads to cell cycle arrest and apoptosis. It has been used effectively for hematologic malignancies including multiple myeloma and mantle cell lymphoma (48).
For npAIR, bortezomib has only been used in adjunct with rituximab, with or without other concurrent immunosuppressive therapy (39,42). In addition to the aforementioned reports, Benson and colleagues discussed a 37-year-old female who presented with photopsias and vision loss over several months. Patient worsened over the next 4 years despite treatment with subtenon triamcinolone acetonide, rituximab (2 infusions of 1,000 mg separated by 2 weeks), azathioprine (150 mg daily, discontinued after 2 weeks due to hepatotoxicity), and methotrexate (25 mg subcutaneous weekly). Patient was then referred to another institution and received the same rituximab regimen along with bortezomib (1.5 mg/m2 subcutaneous, dosed with oral dexamethasone 8 mg, for 3 consecutive weeks; two cycles with an intervening period of 1 week). The patient experienced no improvement and therapy was discontinued, with the patient remaining stable 8 months after treatment cessation (35).
IVIG refers to purified polyclonal serum IgG pooled from ≥10,000 donors, and has been used for the management of various antibody-mediated autoimmune diseases. While its mechanism of action remains incompletely understood, IVIG may reduce the half-life and competitively inhibit the binding of pathogenic autoantibodies, as well as contain anti-idiotype antibodies directed against proinflammatory mediators including activated complement proteins, cytokines, and cell-adhesion molecules (49).
Ferreyra and colleagues reported 3 cases of npAIR treated with IVIG, all of whom showed no response. The first patient (42-year-old female) had first received subtenon methylprednisolone as well as prednisone, cyclosporine and azathioprine without improvement. The second patient (51-year-old female) had also first received cyclosporine, azathioprine, prednisone and intravitreal triamcinolone acetonide. However, she had to stop all oral medications due to adverse effects, and only received 5 months of immunosuppressive therapy. The third patient received IVIG prior to mycophenolate mofetil and intramuscular methylprednisolone acetate, but also showed no improvement (6).
Abazari and colleagues reported three cases of npAIR treated with IVIG after failing to respond to oral prednisone (1 mg/kg) for ≥4 weeks. Only 1 patient who still had normal full-field ERG responses at baseline exhibited improvement on visual field testing and multifocal ERG. The other two patients who had extinguished full-field ERG responses at presentation only experienced subjective visual improvement in photopsias (14).
Plasmapheresis (often used interchangeably with the term therapeutic plasma exchange), refers to removal of blood plasma (along with pathogenic contents including autoantibodies, immune complexes, monoclonal proteins, etc.), usually followed by volume replacement with the patient’s own plasma or a colloidal solution such as 5% albumin (50).
A 31-year-old woman diagnosed with myasthenia gravis at age 17 had undergone thymectomy and prior treatment with IVIG, prednisone, and mycophenolate mofetil prior to developing signs of npAIR at age 23. Her visual acuity worsened to hand motion OU over 2 years, and was not formally diagnosed until age 31, when she had severely depressed visual fields and extinguished ERG responses. Patient was then started on weekly plasmapheresis for over 1 year, which improved her symptoms related to myasthenia gravis but did not augment her visual function. Interestingly, plasmapheresis did not alter the number of circulating antiretinal antibodies (51). In addition, a 41-year-old man with npAIR associated with a renal oncocytoma presented with 2-week history of vision loss OU. Patient showed minimal response to a 5-day course of intravenous methylprednisolone (1 g/day) and five rounds of plasmapheresis. However, after resection of the oncocytoma, he experienced progressive improvement in visual acuity, visual field, OCT, and ERG over 9 months (52).
In contrast, a 35-year-old man diagnosed with autoimmune-related retinopathy and optic neuropathy (ARRON) had worsening vision over 4 years. Visual acuity stabilized after 6 rounds of plasma exchange (53). A 60-year-old woman with npAIR associated with autoimmune cerebellar ataxia also reportedly experienced maintenance of visual function over 21 months after receiving plasmapheresis and IVIG, though details on treatment course and testing parameters were not provided (54).
A 47-year-old woman with ARRON presented with 7-month history of vision loss. She showed initial response to multiple regimens including prednisone, methotrexate, plasmapheresis and IVIG, but her visual function continued to worsen over 3 years. She then underwent nonmyeloablative unmanipulated autologous stem cell transplant, preceded by immune ablation using cyclophosphamide 200 mg/kg and alemtuzumab 20 mg. Her visual acuity and visual field subsequently improved and were stable 2 years post-transplant (55). However, while this report is intriguing, the agents used for immune ablation may also have contributed to the treatment effects (39,56).
Machida and colleagues described a 57-year-old man who underwent positron emission tomography-computed tomography due to rapidly worsening vision over 2 months and clinical suspicion for AIR, which revealed only 18F-fluorodeoxy glucose uptake in the left parotid gland. Histopathology was compatible with a Warthin tumor, and immunohistochemistry demonstrated aberrant expression versus molecular mimicry of recoverin within the neoplasm. In addition, Western blot using the patient’s serum showed immunoreactivity against recombinant human recoverin. Tumor resection was followed by modest improvements in visual acuity and visual fields (57). In contrast, Whitcup and colleagues presented a 62-year-old woman who experienced rapid, progressive vision loss OU over 6 months. Four months prior to the onset of visual symptoms, she had also undergone resection of a Warthin tumor of the left parotid gland. While immunohistochemistry was not performed on the tissue specimen, the patient was found to have elevated serum levels of anti-recoverin (58).
Saito and colleagues reported on a 73-year-old woman who presented with nyctalopia, decreased visual acuity, paracentral scotoma, and recurrent CME OS worsening over 5 years. ERG showed reduced a-waves but normal b-waves OU. Initial Western blot and oncological screening were unremarkable. However, as the patient’s vision also began worsening OD 2 years later in the context of seroconversion to anti-recoverin, repeat systemic workup was performed. Again, no malignancy was found, but patient underwent endoscopic mucosal resection of a colonic polyp. Histopathology was consistent with tubular adenoma with mild atypia, and immunohistochemistry confirmed immunoreactivity against recoverin. The patient experienced significant improvement in CME and visual acuity OS following the procedure, though the authors did not comment on whether this effect was sustained (59).
Finally, as previously discussed, a 41-year-old man had presented with a 2-week history of vision loss OU. Goldmann perimetry and ERG findings were consistent with npAIR. Workup including serologic testing and positron emission tomography-computed tomography revealed multiple antiretinal and anti-optic nerve antibodies [i.e., against 40 kDa (aldolase), 46 kDa (enolase), and 70 kDa (heat shock protein)], as well as a large right renal oncocytoma. Patient did not respond to 5 days of pulse intravenous methylprednisolone (1 g/day) and 5 rounds of plasmapheresis. He then underwent resection of the renal oncocytoma approximately 3 months after presentation, with gradual but significant recovery in visual acuity (20/125 to 20/40 OD; 20/1250 to 20/40 OS), as well as modest improvements in visual field, ERG, and OCT findings (52).
Collectively, these cases provide preliminary support for the idea that benign neoplasms may also induce antiretinal antibodies that lead to npAIR. Of course, benign neoplasms are common incidental findings, and more work is needed to establish a causal relationship.
Given the retrospective nature, small sample sizes, and significant heterogeneity in diagnostic criteria as well as outcome assessment of these studies, the above findings should be interpreted with caution. As with other acquired retinal degenerations, disease severity at presentation and time to treatment are likely important prognostic indicators in npAIR. While some cases are responsive to local/systemic steroids alone, others worsen despite the use of multiple immunosuppressive agents. The limited evidence available also suggests that neither immunosuppression nor plasmapheresis reliably alters the levels or antigenic specificity of antiretinal antibodies. Clearly, additional efforts to define pathogenicity of antiretinal antibodies and standardize measures of treatment response in npAIR are needed.
Corticosteroids and conventional immunosuppressive medications remain first-line treatment for npAIR, given familiarity, safety profile, and time to onset of action. Plasmapheresis and IVIG may be valuable adjunctive therapies. However, the data is sparse, and analysis is further muddled by the fact that patients receiving these modalities often have failed other immunosuppressive agents and/or have more severe disease. Apart from these options, Rituximab is the most widely studied agent for npAIR, with reports of possible efficacy in patients unresponsive to combination regimens including corticosteroids, conventional agents, and IVIG. Consistent with these observations, flow cytometry has further revealed possible aberrations in B-cell maturation in npAIR patients (60). Nevertheless, risks of rituximab as well as immunomodulatory therapy overall should be weighed carefully, given that their effects on npAIR appear to be equivocal.
With regard to other biologics, the current literature suggests that infliximab and adalimumab may not be effective in npAIR refractory to conventional immunosuppressive drugs (6,18,38,41). On the other hand, two recent case reports show that IL-6 antagonists may be useful for npAIR not responsive to conventional agents and other biologics, especially in the setting of retinal vascular leakage and refractory macular edema (17,18). However, it should be noted that both patients exhibited significant angiographic activity, which is unusual for npAIR (5). Interestingly, the only immune profiling study of AIR patients (both npAIR and pAIR) showed that IL-6 and CXCL-9 were elevated among untreated AIR patients compared to treated AIR patients and healthy controls. Moreover, both cytokines were positively associated with disease severity (61).
In summary, the importance of diligent inquiry and investigation to rule out hereditary retinal dystrophies and malignancy cannot be overstated, as npAIR remains a diagnosis of exclusion. Genetic testing should be considered when the presentation is atypical or when there is a high index of suspicion—such as in young patients without established autoimmune disease or individuals with a known family history of retinitis pigmentosa (62). Whether cytokine and immune cell profiling, in conjunction with whole-exome sequencing and epitope mapping, may help identify patients at risk of progression or predict treatment response should be systemically explored (60,61).