
|
表1 本研究在11个家系中发现10个NF1和NF2变异 Table 1. Ten variants in NF1 and NF2 identified in 11 families with different eye conditions in our cohort |
||||||||
|
变异编号 |
外显子 |
碱基改变 |
氨基酸改变 |
家系数 |
ACMG/AMP分类 |
ACMG/AMP证据 |
HGMD |
首次报道 |
|
NF2 (NM_000268.3) |
|
|
|
|
|
|
||
|
M1 |
2 |
c.122G>A |
p.(W41*) |
1 |
P |
PVS1, PS2, PM2 |
DM |
Evans DG, Trueman L, Wallace A, et al. Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating mutations[J]. J Med Genet, 1998, 35(6): 450-455. DOI: 10.1136/jmg.35.6.450 |
|
M2 |
10 |
c.997C>T |
p.(Q333*) |
1 |
P |
PVS1, PM2, PM6 |
DM |
Sestini R, Vivarelli R, Balestri P, et al. Neurofibromatosis type 2 attributable to gonosomal mosaicism in a clinically normal mother, and identification of seven novel mutations in the NF2 gene[J]. Hum Genet, 2000, 107(4): 366-371. DOI: 10.1007/s004390000378. |
|
M3 |
16 |
c.1762C>T |
p.(R588*) |
2 |
P |
PVS1_Moderate, PM2, PM6 |
DM |
Van Hout CV, Tachmazidou I, Backman JD, et al. Exome sequencing and characterization of 49, 960 individuals in the UK Biobank[J]. Nature, 2020, 586: 749-756. DOI: 10.1038/s41586-020-2853-0. |
|
NF1 (NM_000267.3) |
|
|
|
|
|
|
||
|
M4 |
3 |
c.288+1G>A |
/ |
1 |
P |
PVS1, PM2, PM6 |
DM |
A M John, M Ruggieri, R Ferner, M Upadhyaya.A search for evidence of somatic mutations in the NF1 gene[J].J Med Genet. 2000, 37(1):44-9. DOI: 10.1136/jmg.37.1.44. |
|
M5 |
28 |
c.3826C>T |
p.(R1276*) |
1 |
LP |
PVS1, PM6 |
DM |
R A Heim, L N Kam-Morgan, C G Binnie, et al.Distribution of 13 truncating mutations in the neurofibromatosis 1 gene[J]. Hum Mol Genet. 1995, 4(6):975-81. DOI: 10.1093/hmg/4.6.975 |
|
M6 |
29 |
c.3875A>G |
p.(Y1292C) |
1 |
LP |
PS1, PP1, PP3 |
DM |
Ruen Yao, Tingting Yu, Yufei Xu, et al. Clinical Presentation and Novel Pathogenic Variants among 68 Chinese Neurofibromatosis 1 Children[J]. Genes (Basel). 2019, 10(11):847. DOI: 10.3390/genes10110847 |
|
M7 |
31 |
c.4139_4141del |
p.(S1380del) |
1 |
LP |
PVS1, PM2, PM4 |
/ |
本研究 |
|
M8 |
43 |
c.6602delC |
p.(T2201Sfs*11) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
M9 |
45 |
c.6789_6792del |
p.(Y2264Tfs*5) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
M10 |
54 |
c.8003_8006dup |
p.(H2670Vfs*3) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
注:DM:有害变异;P:致病;LP:可能致病。 Note: DM, deleterious mutation. |
||||||||

|
表2. 本研究总11例携带NF1和NF2致病/可能致病变异的不相关先证者的临床表型 Table 2. Clinical manifestations of 11 unrelated probands with P/LP variants in NF1 and NF2
|
|||||||||
|
先证者编号 |
变异编号 |
外显子 |
碱基改变 |
氨基酸改变 |
性别 |
发病年龄 (岁)# |
眼科首诊 |
眼部表型 |
眼外表型 |
|
NF2 (NM_000268.3) |
|
|
|
|
|
|
|
||
|
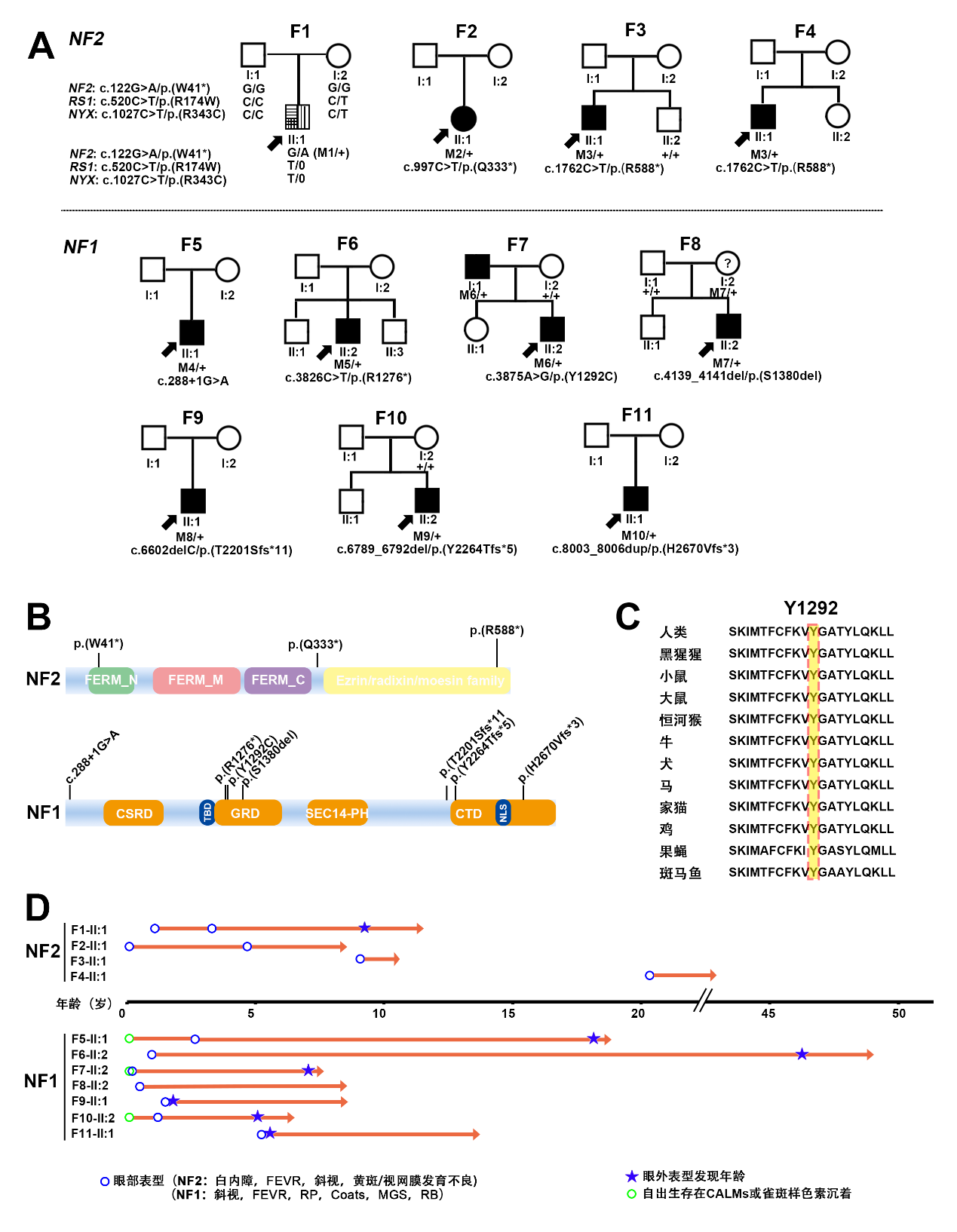
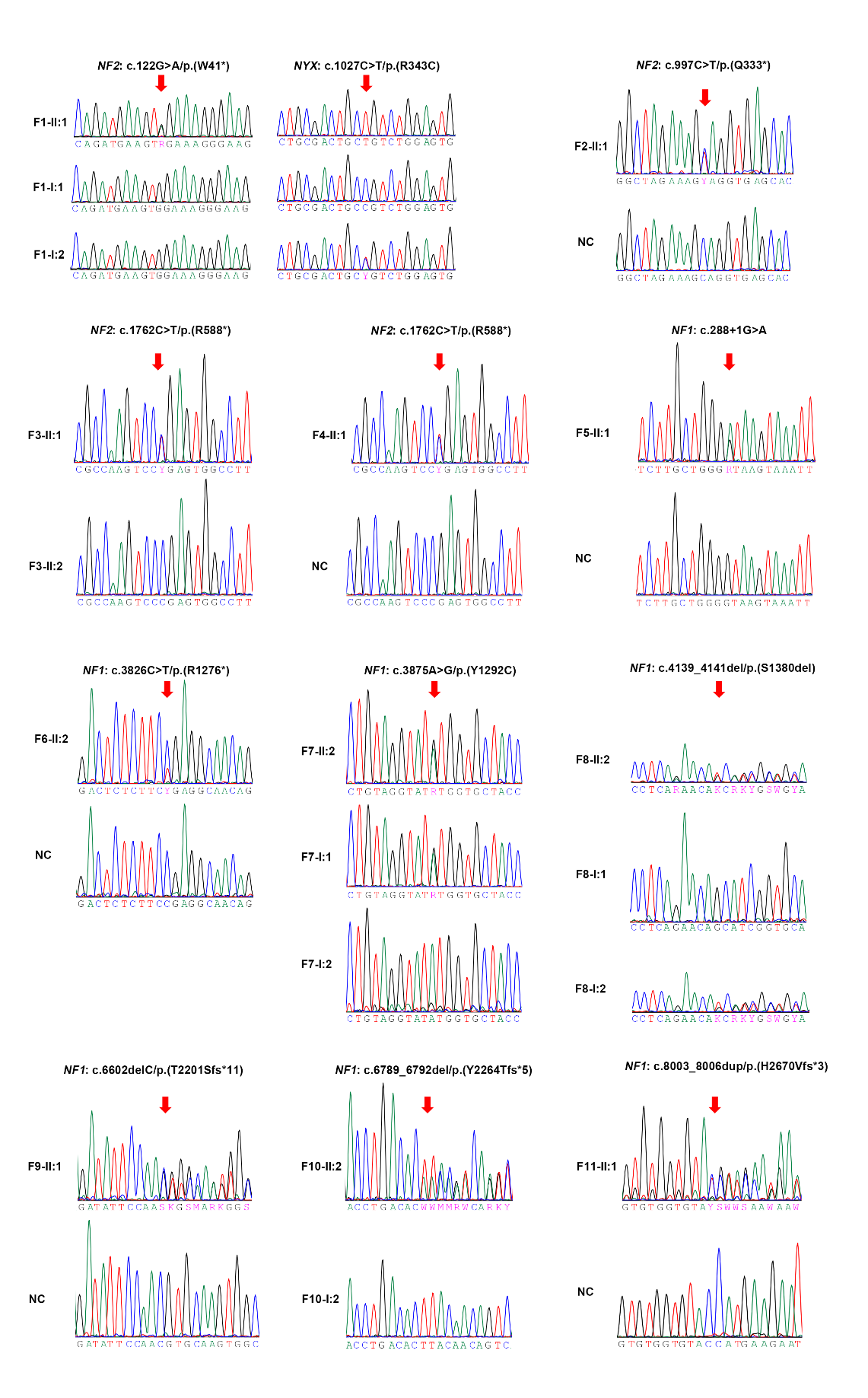
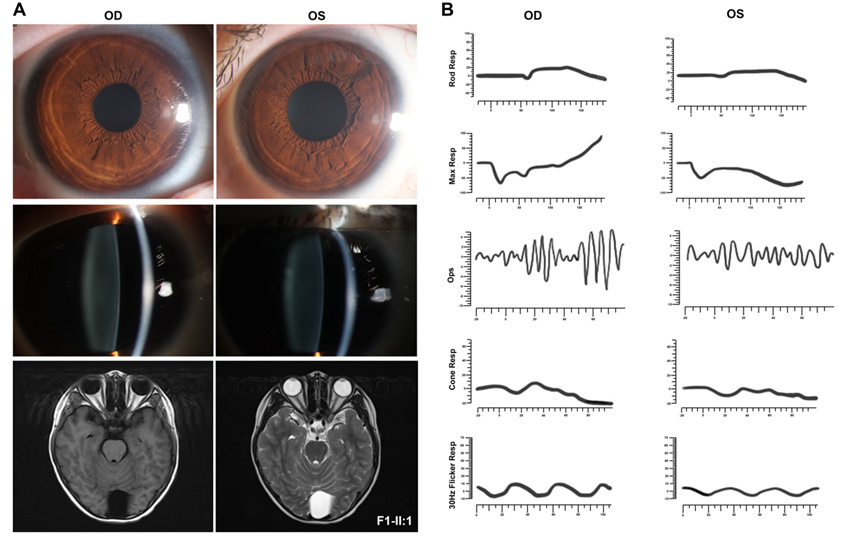
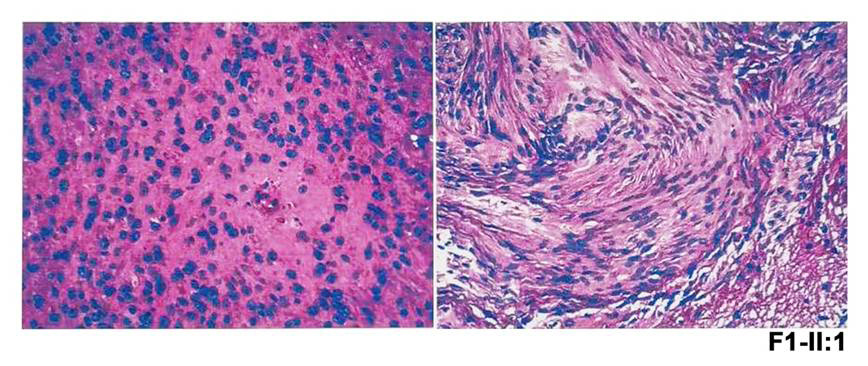
F1-II:1* |
M1 |
2 |
c.122G>A |
p.(W41*) |
男 |
0.25 |
FEVR |
黄斑中央凹反光不明显(右眼);视盘发育不全,黄斑区纤维增殖膜及血管从视盘区拖拽至颞侧中周部视网膜(左眼) |
VS(双侧),脊髓室管膜瘤,多发神经鞘瘤 |
|
F2-II:1 |
M2 |
10 |
c.997C>T |
p.(Q333*) |
女 |
5.6 |
FEVR |
白内障和视网膜脱离(右眼),视网膜前膜及错构瘤(左眼) |
NA |
|
F3-II:1 |
M3 |
16 |
c.1762C>T |
p.(R588*) |
男 |
9 |
RP |
C/D=0.6 |
NA |
|
F4-II:1 |
M3 |
16 |
c.1762C>T |
p.(R588*) |
男 |
33 |
RP |
黄斑发育不良 |
NA |
|
NF1 (NM_000267.3) |
|
|
|
|
|
|
|
||
|
F5-II:1 |
M4 |
3 |
c.288+1G>A |
/ |
男 |
4.5 |
FEVR |
周边视网膜微血管异常 |
CALMs |
|
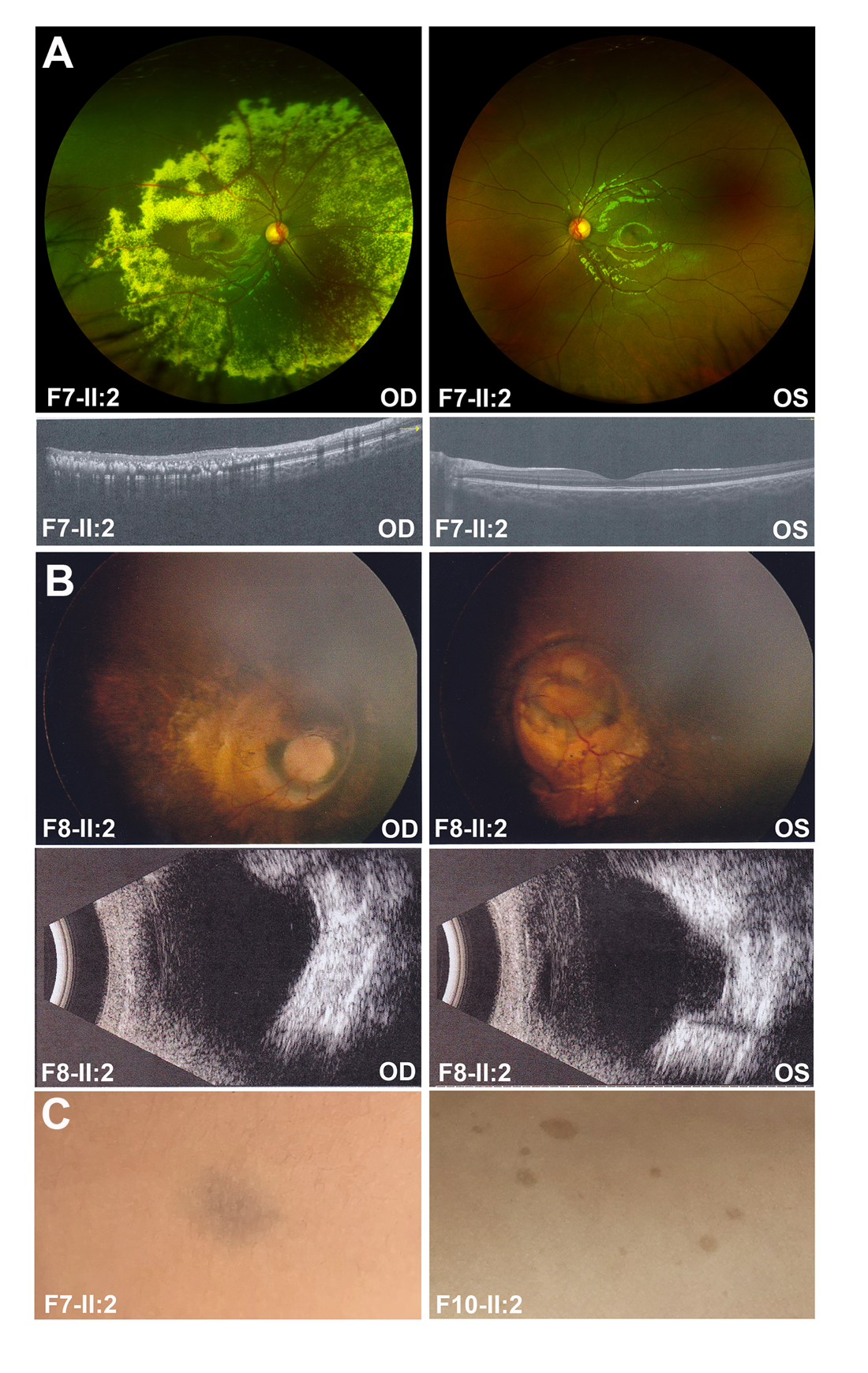
F6-II:2 |
M5 |
28 |
c.3826C>T |
p.(R1276*) |
男 |
儿童期 |
RP |
视网膜色素变性 |
雀斑样色素沉着 |
|
F7-II:2 |
M6 |
29 |
c.3875A>G |
p.(Y1292C) |
男 |
4 |
Coats |
视网膜下渗出 |
雀斑样色素沉着 |
|
F8-II:2 |
M7 |
31 |
c.4139_4141del |
p.(S1380del) |
男 |
0.6 |
MGS |
牵牛花样视盘改变 |
无 |
|
F9-II:1 |
M8 |
43 |
c.6602delC |
p.(T2201Sfs*11) |
男 |
1.3 |
OG |
突眼 |
OG,头部皮下肿块 |
|
F10-II:2 |
M9 |
45 |
c.6789_6792del |
p.(Y2264Tfs*5) |
男 |
1.4 |
斜视 |
斜视 |
CALMs,小脑神经纤维瘤浸润 |
|
F11-II:1 |
M10 |
54 |
c.8003_8006dup |
p.(H2670Vfs*3) |
男 |
6.4 |
OG |
突眼 |
OG |
|
备注:FEVR:家族性渗出性玻璃体视网膜病变;VS:前庭神经鞘瘤;RP:视网膜色素变性;MGS:牵牛花综合征;CALMs:牛奶咖啡斑;OG:神经胶质瘤;NA:未获得。 *F1-II:1基于基因检测及表型分析,最终诊断为NF2相关神经鞘瘤病/XLRS/CSNB1。 |
|||||||||
|
Note: M, male; F, female; FEVR, Familial exudative vitreoretinopathy; ERM, epiretinal membrane; VS, vestibular schwannomas; CHRRPE, combined hamartoma of the retina with or without RPE; RD, retinal detachment; RP, retinitis pigmentosa; MD, macular dystrophy; CALMs, Café-au-lait macules; MGS, Morning glory disc; OG, optic glioma; NA, not available. |
|||||||||


点击右上角菜单,浏览器打开下载














