

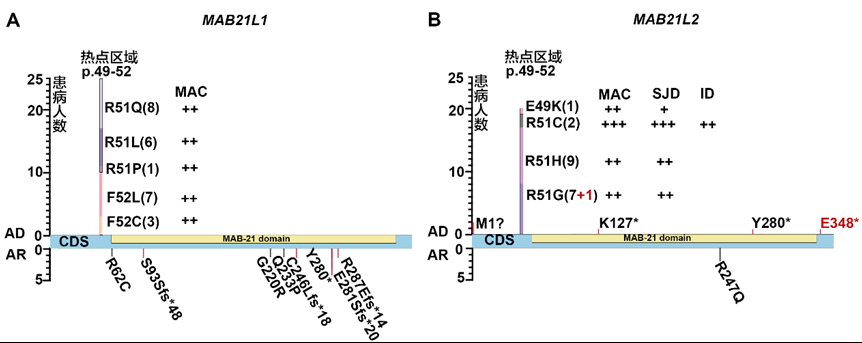

| 致病基因 | 变异 | 蛋白改变 | gnomAD AF | REVEAL | CADD | HGMD | ACMG分级 | ACMG证据 | 文献中ID | 眼部表型 | 其他系统异常 | 文献PMID | ||||||
|
MAC
|
角膜混浊
|
晶体异常
|
眼球震颤
|
骨关节发育异常
|
外生殖器发育不全
|
生长发育迟缓
|
中枢神经系统异常
|
|||||||||||
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS3+PM1+PM2+PP1+PP3+PP4
|
F1-1A
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
33973683
|
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS2+PS3+PM1+PM2+PP1+PP3+PP4
|
F1-1B
|
OU
|
/
|
OU
|
/
|
/
|
/
|
/
|
/
|
33973683
|
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS2+PS3+PM1+PM2+PP1+PP3+PP4
|
F1434-II1
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS2+PS3+PM1+PM2+PP1+PP3+PP4
|
F3413-I1
|
OU
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
36413568
|
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS2+PS3+PM1+PM2+PP1+PP3+PP4
|
Family3-II:2
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[152G>T];[=]
|
p.[R51L];[=]
|
/
|
0.682
|
29.6
|
DM
|
P
|
PS1+PS2+PS3+PM1+PM2+PP1+PP3+PP4
|
Family4-II:2
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[152G>C];[=]
|
p.[R51P];[=]
|
/
|
0.694
|
31
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F592-II:1
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
Family1-II:1
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
Family1-III:1
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
Family2-II:1
|
OU
|
/
|
NA
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F96571-II1
|
OU
|
OS
|
NA
|
/
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F96571-II2
|
OU
|
OD
|
/
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F511-II1
|
OU
|
OU
|
OS
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F511-III1
|
OU
|
/
|
/
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[152G>A];[=]
|
p.[R51Q];[=]
|
/
|
0.542
|
30
|
/
|
P
|
PS2+PM1+PM2+PM5+PP1+PP3+PP4
|
F511-III2
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36413568
|
|
MAB21L1
|
c.[155T>G];[=]
|
p.[F52C];[=]
|
/
|
0.745
|
32
|
/
|
LP
|
PM1+PM2+PP1+PP3+PP4
|
Family5-II:3
|
OU
|
OD
|
OU
|
+
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[155T>G];[=]
|
p.[F52C];[=]
|
/
|
0.745
|
32
|
/
|
LP
|
PM1+PM2+PP1+PP3+PP4
|
Family5-III:1
|
OU
|
/
|
OU
|
/
|
/
|
/
|
/
|
/
|
36892533
|
|
MAB21L1
|
c.[155T>G];[=]
|
p.[F52C];[=]
|
/
|
0.745
|
32
|
/
|
LP
|
PM1+PM2+PP1+PP3+PP4
|
F5531-I:1
|
OU
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
36413568
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-II4
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-II7
|
OU
|
OD
|
+
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-III1
|
OU
|
/
|
+
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-III2
|
OU
|
/
|
+
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-III3
|
OU
|
/
|
OU
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-III4
|
OU
|
/
|
+
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[156C>G];[=]
|
p.[F52L];[=]
|
/
|
0.334
|
24.7
|
/
|
P
|
PS3+PM1+PM2+PP1+PP3+PP4
|
F-III5
|
OU
|
/
|
+
|
+
|
/
|
/
|
/
|
/
|
36446583
|
|
MAB21L1
|
c.[184C>T];
[‐68T>C] |
p.[R62C];[-]
|
0.000038885;
/ |
0.585;/
|
31;/
|
DM?;/
|
B;/
|
PP3+BS2+BS4;/
|
F2-2
|
OS
|
/
|
OD
|
/
|
/
|
/
|
/
|
/
|
33973683
|
|
MAB21L1
|
c.[658G>C];
[*529A>G] |
p.[G220R];[-]
|
0.00000402;/
|
0.446;/
|
26.2;/
|
DM?;/
|
VUS;/
|
PP1+PP3+BS2;/
|
F3-3
|
OU
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
33973683
|
|
MAB21L1
|
c.[698A>C];
[698A>C] |
p.[Q233P];
[Q233P] |
/
|
0.332
|
25.1
|
DM
|
VUS
|
PM2+PP4
|
F2-II3
|
/
|
+
|
/
|
+
|
/
|
+
|
NA
|
CH
|
30487245
|
|
MAB21L1
|
c.[279_286del
ACTGCCCG]; [279_286delAC TGCCCG] |
p.[P95Rfs*47];
[P95Rfs*47] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP4
|
F3-II3
|
/
|
+
|
/
|
/
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[735dupG];
[735dupG] |
p.[C246Lfs*18];
[C246Lfs*18] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP4
|
F1-II3
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
27103078
|
|
MAB21L1
|
c.[840C>G];
[840C>G] |
p.[Y280*];
[Y280*] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F5-IV1
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[840C>G];
[840C>G] |
p.[Y280*];
[Y280*] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F5-VI4
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[840C>G];
[840C>G] |
p.[Y280*];
[Y280*] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F5-VI5
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[841delG];
[841delG] |
p.[E281Sfs*20];
[E281Sfs*20] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F1-VI2
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[841delG];
[841delG] |
p.[E281Sfs*20];
[E281Sfs*20] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F1-VI5
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
NA
|
30487245
|
|
MAB21L1
|
c.[841delG];
[841delG] |
p.[E281Sfs*20];
[E281Sfs*20] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F1-VI6
|
/
|
+
|
/
|
+
|
/
|
+
|
NA
|
NA
|
30487245
|
|
MAB21L1
|
c.[841delG];
[841delG] |
p.[E281Sfs*20];
[E281Sfs*20] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP1+PP4
|
F1-VI7
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L1
|
c.[859delC];
[859delC] |
p.[R287Efs*14];
[R287Efs*14] |
/
|
/
|
/
|
DM
|
P
|
PVS1+PM2+PP4
|
F4-V1
|
/
|
+
|
/
|
+
|
/
|
+
|
+
|
CH
|
30487245
|
|
MAB21L2
|
c.[145G>A];[=]
|
p.[E49K];[=]
|
/
|
0.836
|
32
|
DM
|
LP
|
PM1+PM2+PM6+PP3+PP4+PP5
|
F131-II1
|
OU
|
/
|
/
|
/
|
S
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[151C>T];[=]
|
p.[R51C];[=]
|
/
|
0.868
|
32
|
DM
|
P
|
PS2+PS3+PM1+PM2+PP3+PP4+PP5
|
F676-II1
|
A
|
/
|
/
|
/
|
JC,SL
|
/
|
/
|
ID
|
24906020
|
|
MAB21L2
|
c.[151C>T];[=]
|
p.[R51C];[=]
|
/
|
0.868
|
32
|
DM
|
P
|
PS2+PS3+PM1+PM2+PP3+PP4+PP5
|
F4480-II1
|
A
|
/
|
/
|
/
|
JC,SL
|
/
|
/
|
ID
|
24906020
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
II-7
|
OU
|
/
|
+
|
+
|
JC
|
/
|
/
|
/
|
25719200
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
II-1
|
OU
|
/
|
+
|
+
|
JC
|
/
|
/
|
/
|
25719200
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
II-5
|
OU
|
/
|
+
|
+
|
JC
|
/
|
/
|
/
|
25719200
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
III-1
|
OU
|
/
|
+
|
+
|
JC
|
/
|
/
|
/
|
25719200
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
III-2
|
OU
|
/
|
+
|
+
|
JC
|
/
|
/
|
/
|
25719200
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
P
|
PS1+PM1+PM2+PP3+PP4+PP5
|
F1P1-III1
|
OU
|
/
|
/
|
/
|
JC
|
/
|
/
|
/
|
35929935
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
P
|
PS1+PM1+PM2+PP3+PP4+PP5
|
F1P2-II1
|
OU
|
/
|
/
|
/
|
JC
|
/
|
/
|
/
|
35929935
|
|
MAB21L2
|
c.[151C>G];[=]
|
p.[R51G];[=]
|
/
|
0.851
|
29.8
|
DM
|
P
|
PS1+PS3+PM1+PM2+PP3+PP4+BS4
|
F1-II2
|
OU
|
OS
|
NA
|
OU
|
JC
|
/
|
/
|
/
|
本文
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-III1
|
OU
|
/
|
/
|
/
|
JC
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-I1
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-II1
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-II5
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-II6
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-III2
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-III10
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-III11
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[152G>A];[=]
|
p.[R51H];[=]
|
/
|
0.879
|
32
|
DM
|
LP
|
PM1+PM2+PM5+PP1+PP3+PP4+PP5
|
F1463-III12
|
+
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[1A>C];[=]
|
p.[M1?];[=]
|
/
|
/
|
/
|
DM
|
LP
|
PM2+PM4+PP4+PP5
|
P40
|
OU
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
32737437
|
|
MAB21L2
|
c.[1A>C];[=]
|
p.[M1?];[=]
|
/
|
/
|
/
|
DM
|
LP
|
PM2+PM4+PP4+PP5
|
P40-father
|
OU
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
32737437
|
|
MAB21L2
|
c.[379A>T];[=]
|
p.[K127*];[=]
|
/
|
/
|
/
|
DM
|
VUS
|
PM2+PM4+PP4
|
16–1
|
U
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
36192130
|
|
MAB21L2
|
c.[840C>G];[=]
|
p.[Y280*];[=]
|
/
|
/
|
/
|
DM
|
VUS
|
PM2+PM4+PP4
|
P14DG0332
|
U
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
29450879
|
|
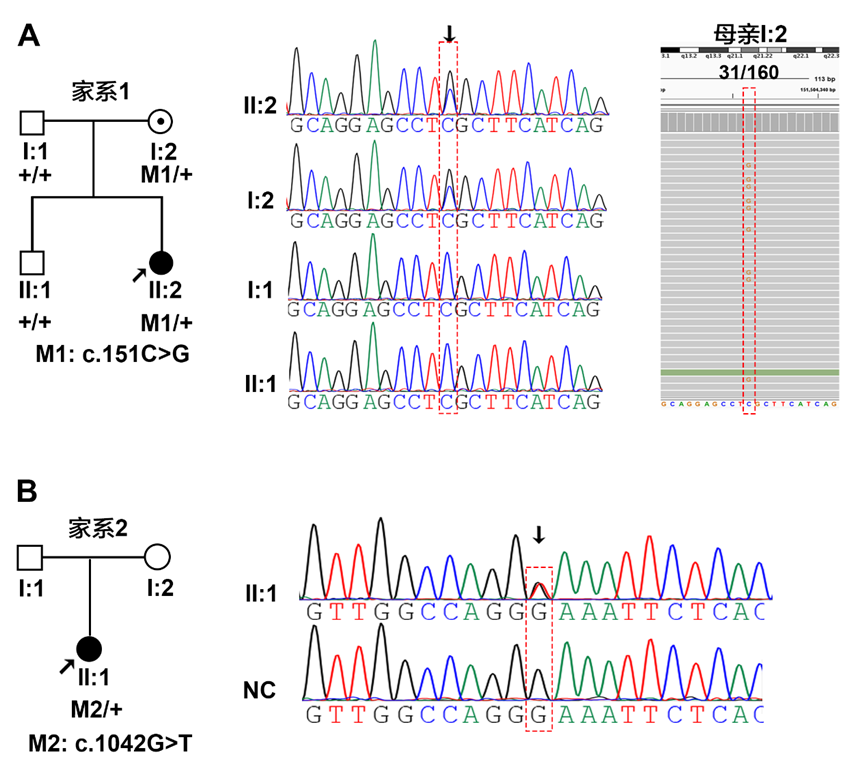
MAB21L2
|
c.[1042G>T];[=]
|
p.[E348*];[=]
|
/
|
/
|
/
|
/
|
LP
|
PM2+PM4+PM6+PP4
|
F1-I1
|
OS
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
NA
|
本文
|
|
MAB21L2
|
c.[740G>A];
[740G>A] |
p.[R247Q];
[R247Q] |
/
|
0.826
|
33
|
DM
|
VUS
|
PM2+PP3+PP4+PP5
|
F4468-II1
|
OU
|
/
|
/
|
/
|
/
|
/
|
/
|
/
|
24906020
|
|
MAB21L2
|
c.[740G>A];
[740G>A] |
p.[R247Q];
[R247Q] |
/
|
0.826
|
33
|
DM
|
VUS
|
PM2+PP3+PP4+PP5
|
F4468-II2
|
OU
|
/
|
/
|
/
|
MSL
|
/
|
/
|
/
|
24906020
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 注:本文报道的变异用橙色标记。 F: 家系; OU:双眼 ;OD:右眼;OS:左眼;U: 单眼; “+”: 有该表型; “/”: 没有该表型;A: 无眼球;CH:小脑发育不全;ID:智力障碍;JC:关节屈曲挛缩;S:并指;SL:四肢短缩;MSL:轻微四肢短缩;NA: 未做相关检查;HGMD:人类基因突变数据库;ACMG:美国医学遗传和基因组学学会指南;UTR:非翻译区;DM:有害突变;D:有害的;T:耐受的;P:致病的;LP:可能致病的;VUS:意义不明确的;B:良性的。 | ||||||||||||||||||
| Notes: The variants reported in this article were marked in orange. F: family; OU: both eyes; OD: right eye; OS: left eye; U: unilateral eye; "+": with this phenotype; "/": without this phenotype; A: anophthalmia; CH: cerebellar hypoplasia; ID: intellectual disability; JC: joint contracture; S: syndactylia; SL: shortened limbs; MSL: mildly shortened limbs; NA: not available; HGMD: The Human Gene Mutation Database; ACMG: American College of Medical Genetics and Genomics; UTR: untranslated region; DM: damaging mutations; D: damaging; T: tolerant; P: pathogenic; LP: likely pathogenic; VUS: variants uncertain significance; B: benign. | ||||||||||||||||||


点击右上角菜单,浏览器打开下载














