Abstract: Hereditary, metabolic and toxic optic neuropathies cause bilateral, central vision loss and therefore can result in severe impairment in visual function. Accurate, early diagnosis is critical, as nutritional and toxic optic neuropathies may be reversible if identified early, and diagnosis of hereditary optic neuropathies can prevent unnecessary invasive workup, provide prognostic information, and allow for effective genetic counseling. Optical coherence tomography (OCT) is a valuable tool that aids in the diagnosis and prognostication of optic neuropathies as it allows for quantification of changes in the retinal ganglion cells (RGCs) and retinal nerve fiber layer (RNFL) over time. We review the characteristic clinical presentations of hereditary, metabolic and toxic optic neuropathies, with an emphasis on OCT findings.
Most metabolic, toxic and hereditary optic neuropathies are thought to arise from mitochondrial dysfunction. They present with similar symptoms, including painless central or cecocentral scotomas, reduced visual acuity and dyschromatopsia. Vision changes are typically bilateral, or unilateral with sequential involvement of the second eye. The onset can be sudden or insidious, depending on the etiology, and visual outcomes can range from complete recovery to no light perception vision. Exam characteristically shows selective thinning or pallor of the temporal aspect of the optic nerve commensurate with the extent of optic nerve injury, especially in the setting of prolonged disease.
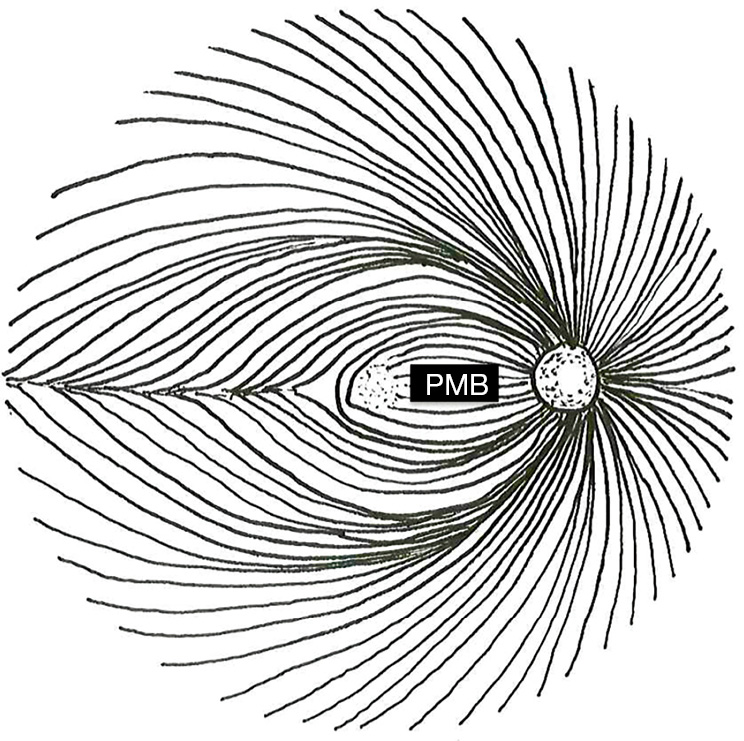
These characteristics stem from preferential damage to the papillomacular bundle (PMB) in mitochondrial optic neuropathies. Retinal ganglion cell (RGC) axons course over the anterior surface of the retina in the retinal nerve fiber layer (RNFL) in distinct geographical bundles to converge on the optic nerve head. The PMB carries fibers from the macula to form the temporal aspect of the optic nerve. In contrast, the superior and inferior polar regions of the nerve are formed by fibers arising superior and inferior to the horizontal raphe temporal to the fovea, while the nasal optic nerve encompasses fibers from the nasal periphery (Figure 1). Therefore, damage to the PMB impacts macular function and results in temporal optic nerve pallor on fundus examination and RNFL thinning with OCT.
Several atypical features of the axons within the PMB place them under increased metabolic stress. They are unmyelinated and require large amounts of energy to propagate action potentials relative to the saltatory conduction present in myelinated nerves. The PMB also carries a relatively high proportion of axons arising from small-caliber, rapid-firing parvocellular RGCs (1-4). Small-caliber axons are thought to be more susceptible to metabolic stress, as models of mitochondrial optic neuropathy based on axon caliber accurately predict nerve fiber loss in pathologic sections from patients with mitochondrial optic neuropathy (2). This is attributed to the axon’s lower volume-to-surface-area ratio (which equates to having fewer mitochondria available to maintain membrane potentials). Finally, as the nerve fibers exit the eye, they make a sharp turn to pass through the lamina cribrosa. This configuration represents a “choke point” of restricted axoplasmic flow in which there are increased metabolic demands for intracellular transport. Histologic sections of the optic nerve demonstrate a high concentration of mitochondria in this area (5). Overall, the PMB appears to be more susceptible to damage in the setting of mitochondrial dysfunction due to the energetic demands of small-caliber, unmyelinated nerve fibers traveling in a physically constricted space. This helps explain why mitochondrial dysfunction (which should be present in every cell in the body) can selectively affect the optic nerve in general and the PMB fibers in particular.
Mitochondrial optic neuropathies can sometimes be difficult to distinguish from other causes of optic nerve injury including glaucoma, inflammatory disorders, ischemia, compression, trauma or malignancy. Often a thorough history and exam helps to narrow the differential diagnosis. Ancillary testing including visual fields, fundus autofluorescence and optical coherence tomography (OCT) can be helpful in equivocal cases, in addition to blood work and imaging. OCT is a non-invasive, relatively affordable imaging modality that allows for high-resolution, quantitative visualization of the optic nerve and macula. This technology allows for quantification of axonal and neuronal loss, which can serve as a valuable marker for progression of visual loss, as well as a predictor for visual recovery with treatment.
Several OCT modalities are helpful in the setting of mitochondrial optic neuropathies. Assessment of the RNFL with OCT provides objective measurement of sectoral swelling or atrophy of the optic nerve, which can be followed over time to track improvement or progression. Macular OCT with GC-IPL segmentation allows for quantification of the ganglion cell layer and inner plexiform layer (IPL). In the setting of acute injury, GC-IPL analysis is particularly useful, as GC-IPL thinning may be apparent while RNFL thinning is masked by active swelling or distention of the nerve fibers. In other conditions, including optic neuritis, GC-IPL thinning can precede measurable RNFL thinning even in the absence of swelling. This surrogate marker of neuronal integrity can also provide prognostic information and be followed over time to track progression. In patients with central scotoma, macular OCT also provides visualization of the outer retina, which is helpful in distinguishing between disease processes effecting the retina and optic nerve. A few early studies of OCT-angiography in hereditary optic neuropathy demonstrate detectable changes prior to the onset of RNFL thinning, suggesting that it may be a helpful technique as it becomes more widely available (6-8). Different hereditary, toxic and metabolic optic neuropathies manifest somewhat different findings on OCT, as discussed below.
First described by Theodore Leber in 1871, LHON often causes sequential bilateral vision loss in young adults. The majority of cases (90–95%) are attributed to mitochondrial DNA mutations at position G11778A, T14484A, and G3460A, which effect NADH dehydrogenases (ND4, ND6, and ND1, respectively) (9,10). These dehydrogenases are complex I proteins that play a critical role in oxidative phosphorylation, neutralizing free radicals and regulating apoptosis (10). Prevalence varies somewhat between different populations, with the prevalence in Europe being roughly 1:30,000. LHON exhibits variable penetrance, with 50% of male mutation carriers and 10% of female carriers developing vision loss. The typical age of onset ranges from 15 to 35, but cases have been reported in all decades of life (9).
Clinically, LHON is characterized by the sequential, painless onset of dense cecocentral scotomas with dyschromatopsia. Visual acuity is typically reduced to 20/200 or less, and the fellow eye is almost always affected within the first year, most commonly within 6–8 weeks. Vision loss typically progresses for up to 6 months and then plateaus. Some visual improvement is possible, particularly in patients carrying the 14,484 mutation, for which a 37–71% chance of partial visual recovery has been reported, relative to 4% for the other mutations (9). Fundus exam at the time of onset typically shows thickening of the RNFL, hyperemia, peripapillary telangiectasias and mild tortuosity, although these findings are not always present. Fluorescein angiography characteristically shows the absence of leakage at the optic nerve head, which can help in distinguishing this process from other optic neuropathies.
Several longitudinal studies of patients with LHON have described characteristic RNFL changes on OCT. Asymptomatic carriers have statistically significant thickening of the temporal RNFL relative to age-matched controls, and a trend towards inferior RNFL thickening (11). This early thickening has been attributed to impairment of axoplasmic flow and likely represents early or baseline stress to the nerve fibers. At the time of symptom onset, there is typically predominant thickening of the superior and inferior RNFL, with normalization of the temporal RNFL as it begins to atrophy (12). These changes correspond to the appearance of disc edema, hyperemia and microvascular change on fundus exam. After 3 months, temporal thinning can be appreciated, and by 9 months superior and inferior thinning is apparent as well (13).
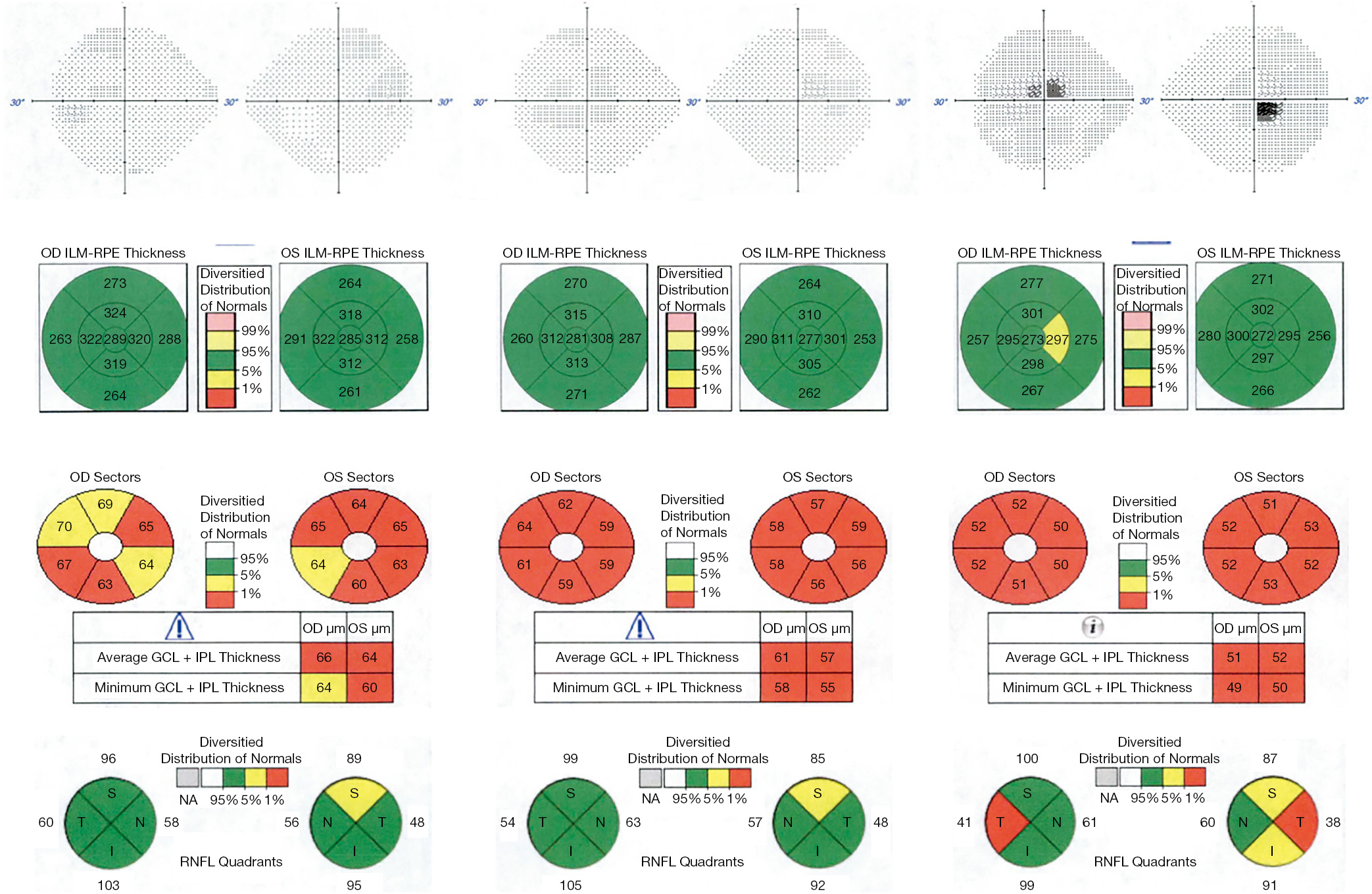
Progressive changes in the macula have been characterized in more recent case series of LHON patients. Macular OCT shows thinning of the inner macular ring and nasal peripheral macula in the first 3 months after symptom onset. This thinning then progresses to involve the temporal macula over the next 3 months, and by 12 months, diffuse macular thinning is evident, with average macular thickness of 251 μm compared with an average of 281 μm in controls (14). GC-IPL analysis appears to be even more sensitive to early changes in LHON. A small longitudinal case series of 4 patients demonstrated thinning of the GC-IPL in the nasal macula prior to symptom onset. In the first three months after the onset of symptoms, the GC-IPL thinning progressed to involve the inferior, then superior and temporal sectors with diffuse thinning of the average GC-IPL to 63 μm at 3 months. This thinning continued to progress to 57 μm at 6 months, then stabilized (7). See Figure 2 for a discussion of a case with typical progressive changes on OCT.
Recent case reports have indicated that changes in the radial peripapillary capillary (RPC) network can be appreciated in patients with LHON using OCT-angiography (15-17). This has been more thoroughly explored in a case series of 22 patients at different stages in disease progression. Capillary dropout in the RPC network was noted, with decreased vascular density occurring temporally in early disease stages, followed by inferior and superior capillary dropout, then diffuse dropout in chronic disease. The changes in vascularity corresponded with GC-IPL thinning, and preceded RNFL thinning (6). As OCT-angiography becomes more widely available, it may prove a useful correlate to GC-IPL in tracking disease progression and response to therapy, particularly in the early stages of the disease.
Given the sequential vision loss in LHON, there is a small window of opportunity in which prompt diagnosis may allow intervention prior to loss of vision in the fellow eye. A randomized, double-blinded, placebo-controlled trial demonstrated that idebenone 900 mg/d results in mild improvement of visual acuity outcomes in patients with discordant visual acuities at the onset of treatment, particularly in patients with the 11,778 and 3,460 mutations (18). Idebenone is not currently approved for treatment of LHON in the United States or Canada, but has been approved in the European Union for this purpose. Gene therapy is another emerging treatment option for LHON. Stage I/II clinical trials have been completed in 15 patients with the 11,778-mutation demonstrating a favorable safety profile, and stage III trials (RESCUE and REVERSE) are ongoing (19). Patients are likely to have maximal benefit from these treatment options with early diagnosis. Quantification of axonal and neuronal loss with OCT in general, and GC-IPL analysis in particular, can provide critical early diagnostic information for LHON, which will assist in identifying patients in the early stages of disease who may benefit the most from intervention as more treatments become available.
ADOA, first described by Paul Kjer in 1957, typically presents earlier than LHON, with most patients developing symptoms in the first decade of life. Vision loss is bilateral, simultaneous and gradual in onset, and very slowly progressive over the course of decades. Patients most commonly exhibit reduced visual acuity, cecocentral scotomas and blue/yellow dyschromatopsia. The majority of patients retain visual acuity of 20/200 or better, with one third retaining 20/60 or better, and there is a high degree of variability in penetrance and expressivity within families (20). While cecocentral scotomas are most common, these patients can also present with bilateral superotemporal visual field loss that does not respect the vertical midline (21). The optic disc characteristically exhibits temporal pallor or excavation, which can progress to diffuse pallor later in the course of disease (22,23). Prevalence ranges from 1:10,000 to 1:50,000 worldwide, with the highest incidence in Denmark (24-26).
Mutations in the OPA1 gene are most frequently associated with ADOA, although cases attributed to OPA3 mutations have been reported as well (24). Over 100 different OPA1 mutations have been implicated in ADOA, and OPA1 mutations are identified in 32% to 89% of cases. Both missense and haplo-insufficiency mutations have been identified, with more severe atrophy associated with missense mutations (27). OPA1 encodes a dynamin-related GTPase that localizes to the mitochondrial inner membrane, and plays an important role in mitochondrial fusion, oxidative phosphorylation and apoptosis. Therefore, although ADOA follows a Mendelian inheritance pattern, it behaves like a mitochondrial optic neuropathy. Apoptosis of RGCs is a critical part of normal development, with an estimated reduction in RGC numbers from 2.2–2.5 to 1.5–1.7 million over the course of development in utero (28). Patients with vision loss and confirmed OPA1 mutations have optic discs that are smaller than those in age-matched controls, and more severe phenotypes are associated with smaller discs, suggesting that greater rates of apoptosis in RGCs may drive this disease (29,30). Histologic studies of patients of with ADOA are consistent with RGC dropout, demonstrating fibrosis and cell loss of the RGC layer, with associated myelin loss and increased fibrosis in the optic nerve and lateral geniculate nucleus (LGN) (31,32).
Analysis of RNFL thickness using OCT has consistently demonstrated a severe reduction in average RNFL thickness in patients with ADOA compared with healthy controls. This thinning affects the entire optic nerve, but like other mitochondrial-related optic neuropathies is most prominent temporally, with multiple cross-sectional studies demonstrating thinning that is most pronounced temporally and inferiorly, then superiorly, and least pronounced nasally (27,30,33-36). This characteristic pattern can help distinguish ADOA from other optic neuropathies presenting in childhood including glaucoma, inflammatory optic neuropathies and compressive lesions. RNFL thinning occurs with aging in the general population. Curiously, the rate of RNFL thinning from the first to eighth decade of life appeared comparable in patients with ADOA compared with healthy controls, as assessed in two cross-sectional studies (33,34). This suggests that pathologic RGC is established very early in life, perhaps even prenatally. Clinically, we have also observed that patients with ADOA tend to perform better on automated perimetry than patients with other optic neuropathies and comparable RNFL thinning, which could reflect improved cortical adaptation.
Noninvasive imaging of specific retinal layers in the setting of ADOA has been undertaken with macular OCT (37,38). This approach demonstrates thinning of the inner retinal layers (GCL, IPL), while the outer retinal layers (INL, OPL, ONL, ellipsoid zone, RPE) appear intact (27,37). In particular, GC-IPL thinning is seen early in the course of disease, and shows more pronounced changes than the RNFL (27,30). Therefore, macular OCT with GC-IPL analysis may helpful in the setting of childhood-onset vision loss, to distinguish between ADOA and retinal dystrophies. Correlations between visual acuity and OCT measures including RNFL thinning and GC-IPL thinning have been described (8,27,39). However, OCT is not used routinely for predicting the degree of subsequent vision loss.
Recent case-control studies using OCT-angiography in patients with ADOA have demonstrated reduced density of the temporal RPC network and macular superficial capillary plexus, which supply the RGCs. The deep capillary plexus and the foveal avascular zone were unaffected (8,40). These studies yielded conflicting results on the correlation between RPC density and visual acuity. There was a stronger correlation between visual acuity and GC-IPL thickness, suggesting that GC-IPL analysis may be the most useful OCT modality currently available for objectively tracking vision-affecting changes over time. It is unclear if the vascular changes noted are secondary to RGC loss, or if capillary dropout plays a role in the pathogenesis of ADOA. Future studies might explore OCT-angiography in younger patients or carriers to better define the progression of vascular changes early in the course of disease.
Optic neuropathy can occur in the context of other more rare mitochondrial syndromes, which have some overlapping genetic risk factors. Wolfram Syndrome, also known as DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), has been attributed to mutations in WFS1 and WFS2, but there is also increased risk in carriers of mitochondrial mutations associated with LHON. Behr’s syndrome (optic atrophy, ataxia, mental retardation, urinary incontinence, pes cavus) is associated with OPA3 in some pedigrees. Optic atrophy has also been described as a feature of Friedrich’s ataxia, specific forms of spinocerebellar ataxia (SCA1 and SCA3), mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), and chronic progressive external ophthalmoplegia (CPEO). A high level of suspicion for optic neuropathy should be maintained when patients diagnosed with these syndromes present with vision changes, particularly if the clinical presentation, exam and ancillary testing are consistent. In these cases, OCT can be a valuable adjunct to the examination, allowing more precise assessment of optic atrophy and progression over time, particularly in patients who cannot reliably perform psychophysical tests such as visual acuity and perimetry.
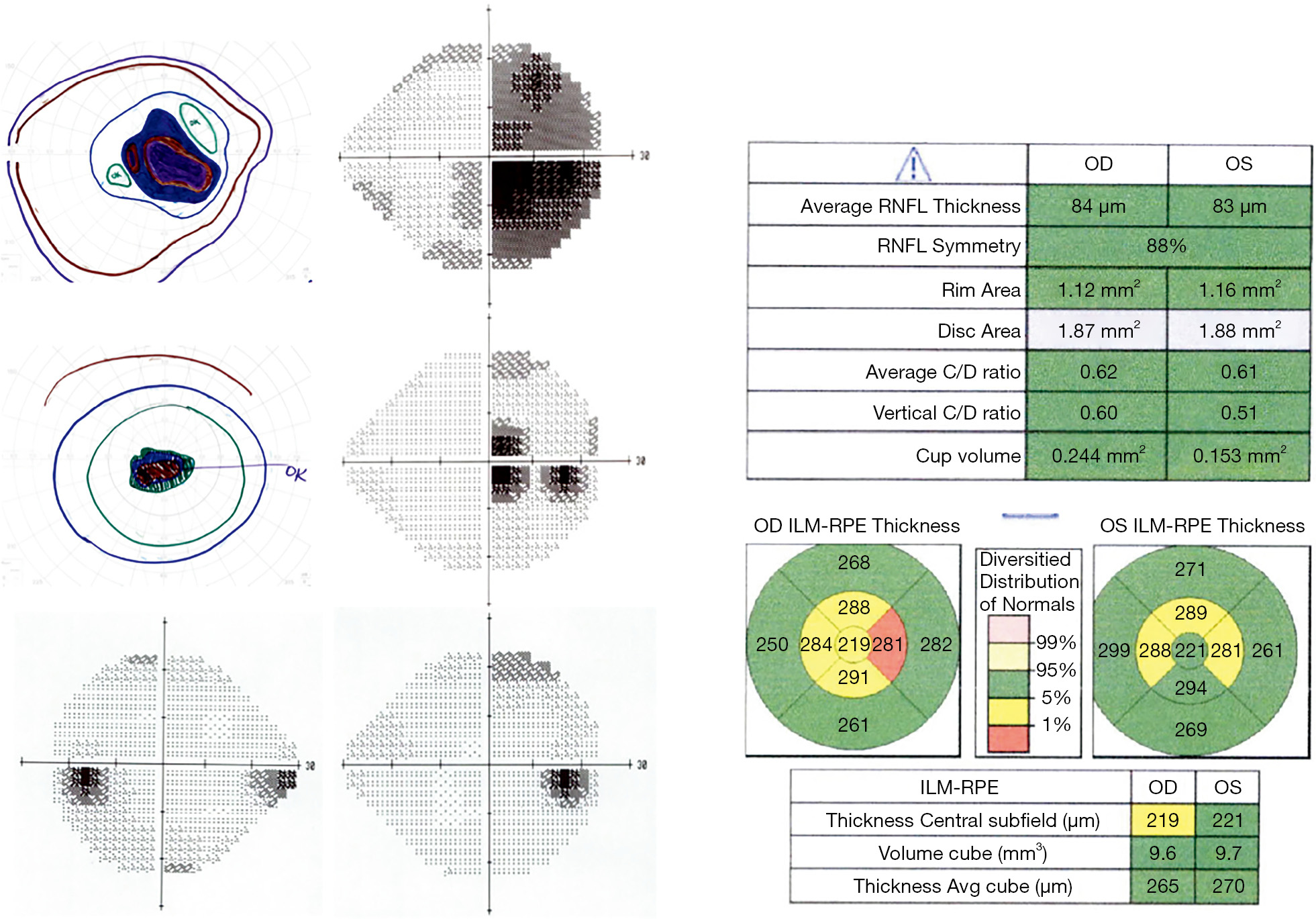
Ethambutol is most commonly used in the treatment of Mycobacterium tuberculosis and Mycobacterium avium complex infections. Mitochondrial toxicity is thought to result from its activity as a metal chelator, as local depletion of copper at the mitochondria inhibits cytochrome C oxidase, slows oxidative phosphorylation and triggers apoptosis (41). Ethambutol-induced optic neuropathy occurs in a dose-responsive fashion. Under current recommended dosing regimens, 1% of patients develop toxicity, presenting with bilateral central vision loss, along with diminished color vision and contrast sensitivity (42,43). A subset of patients do not present with central or cecocentral scotomas, but instead develop junctional scotomas (Figure 3) or bitemporal hemianopsia that does not respect the vertical midline (44). The vision loss typically progresses for 1–2 months after discontinuing the medication, then gradually improves over the subsequent 6 months with partial or full recovery of vision.
Several small case series have demonstrated that OCT of the RNFL is often normal near the time of symptom onset, with progressive thinning over the subsequent several months. This thinning is diffuse, with decreased average RNFL thickness (by 19–48%), but is most prominent temporally (with 19–72% thinning) (45-49). A small case series evaluating full thickness macular OCT demonstrated thinning throughout the inner macula, and outer nasal macula, which corresponds to the area of the PMB (50). The utility of GC-IPL thickness analysis as compared to RNFL thickness has also been assessed in multiple case series, which suggest that thinning of the GC-IPL appears earlier than RNFL thinning, and is a better predictor of visual outcome (51-53).
Early identification of toxicity is critical, as partial visual recovery is possible with discontinuation of the medication. Current screening recommendations for ethambutol-induced optic neuropathy include regular monitoring of visual acuity, color vision and visual fields (54). The role for OCT in screening asymptomatic patients is unclear. As detailed above, case reports have demonstrated that OCT detects minimal change in the RNFL at the time of symptom onset. Furthermore, prospective studies evaluating OCT of the RNFL have shown mixed results, with one study indicating an increase in average RNFL thickness, while two other studies reported sectoral thinning in a subset of patients (55-57). Assessment of GC-IPL thickness may be more promising as a screening tool, as GC-IPL changes have been documented prior to RNFL changes, and have been correlated with visual acuity as well.
Methanol ingestion is associated with metabolic acidosis and optic neuropathy. The neuropathy may be secondary to demyelination of the optic nerve posterior to the lamina cribrosa (58). The methanol metabolite formic acid is a known inhibitor of mitochondrial cytochrome C oxidase, which is essential for oxidative phosphorylation and likely has a direct toxic effect on retinal nerve fibers as well (59). While accidental or intentional ingestion does occur in isolated cases, there have periodically been outbreaks of toxicity associated with commercial distribution of tainted ethanol, which has allowed for longitudinal studies of groups of patients with methanol intoxication.
Patients with methanol intoxication experience bilateral, acute onset, decreased visual acuity (which can be as severe as no light perception), visual field changes and photophobia, followed by gradual improvement over the subsequent weeks. In a retrospective study of 122 patients impacted by an outbreak of methanol poisoning in Ahmedabad, India in 2009, disc edema was noted at the time of presentation in 42% of patients, and optic nerve pallor in 20% of patients after 3 months (60). Visual prognosis depended on the visual acuity and serum pH at presentation, with a serum pH <7.3 associated with an increased risk of chronic RNFL thinning (OR 11.65) (60,61). Chronic changes were also noted in 42 patients involved in a mass methanol poisoning in the Czech Republic in 2012, with, 40% of patients developing chronic visual sequela. These patients also demonstrated RNFL thinning at 5 months, while only rare borderline changes were noted in patients who returned to baseline vision (61,62). Visual acuity remained relatively stable over the 4-year follow-up period, although there was some progression in RNFL thinning in severe cases. GC-IPL analysis has not been conducted in a large study group, but a case study of a single patient with persistent small cecocentral scotomas reported normal OCT RNFL, with mild thinning of the nasal macula GC-IPL at 1 month, which was more prominent at 8 months (63).
The presence or absence of RNFL and GC-IPL thinning on OCT may be helpful in counseling patients on visual prognosis, particularly several months after ingestion. As in other forms of mitochondrial optic neuropathy, changes in the GC-IPL may precede detectable RNFL changes. No intervention has been definitively shown to improve prognosis, although steroids and erythropoietin have both been reported anecdotally (64,65).
Amiodarone is an anti-arrhythmic that is associated with optic neuropathy in 2% of patients. Amiodarone-associated optic neuropathy has been attributed to lamellar inclusion bodies within the optic nerve oligodendrocytes secondary to lysosomal dysfunction, rather than mitochondrial dysfunction (66). The average time to onset in a meta-analysis of 296 patients was 6 to 9 months after initiation of therapy (67). Classically, patients experience insidious, mild, bilateral vision loss (to 20/30 on average). Disc edema is documented in 85% of patients. However, the clinical presentation can vary. Some patients develop acute, or more severe loss of vision, and 35% present with unilateral symptoms and disc edema (68). These unilateral cases can be difficult to distinguish from NAION, particularly as patients on amiodarone are likely to have vasculopathic risk factors for NAION. The key distinguishing feature is that disc edema in NAION typically resolves after several weeks, while the edema associated with amiodarone toxicity persists for several months. Serial measurements of the RNFL using OCT are essential in equivocal cases.
There are no formal recommendations for screening intervals, although a baseline exam is recommended by the Heart Rhythm Society (67). Given the typical onset within the first year of therapy, it has been suggested that a baseline exam, with interval exams during the first year, and annual exams thereafter is a reasonable approach (68). In patients who develop edema or vision changes, serial OCT of the RNFL may be helpful for following the resolution of edema and distinguishing between amiodarone-associated optic neuropathy and NAION. Improvement in vision was noted in 58% of patients after discontinuing amiodarone, so accurate diagnosis and consultation with the patient’s cardiologist is essential, particularly in more severe cases (67).
Linezolid is an oxazolidinone antibiotic that inhibits mitochondrial protein synthesis, and can cause optic neuropathy after prolonged use for greater than 1 month (69). Patients typically present with reduced visual acuity and color vision, with cecocentral scotomas and bilateral disc edema (70). Dramatic partial, if not complete, improvement is obtained with discontinuation of the medication. Temporal pallor is sometimes seen as a late finding. Only a small number of case reports have included OCT imaging, which shows diffuse RNFL thickening in the acute stage (70-72). Given the nonspecific OCT findings, a high level of suspicion in patients presenting with typical symptoms and prolonged use of linezolid (longer than 1 month) is essential for diagnosis. Prompt diagnosis is critical, as vision improves with withdrawal of linezolid. Steroid treatment has been attempted, but appears to worsen rather than improve symptoms (73,74).
Deficiencies of cobalamin (vitamin B12), thiamine (vitamin B1), folate and copper have been associated with optic neuropathy (75-86). Symptoms are typical of mitochondrial optic neuropathy and include decreased visual acuity, dyschromatopsia and cecocentral scotoma. Patients often have an associated peripheral neuropathy, or macrocytic anemia in the setting of B12 or folate deficiency, but the optic neuropathy can occasionally be isolated. In the developed world, B12, thiamine and copper deficiency occur most often in the setting of poor absorption secondary to gastrointestinal disease or gastric bypass surgery. Folate deficiency has been associated with poor diet, alcoholism and long-term use of low-dose methotrexate. There have also been historical reports of outbreaks of optic neuropathy in populations suffering from nutritional strain due to sociopolitical factors, most recently the Cuban epidemic neuropathy from 1991–1993 (87).
The most comprehensive assessment of OCT findings in metabolic optic neuropathy is case series of 45 patients with B12 deficiency, which showed thinning of the average and temporal RNFL (88). Several case studies have been conducted on patients with copper deficiency who presented one to three years after gastric bypass surgery (75-78). Temporal pallor, with associated RNFL thinning was noted, along with GC/IPL thinning. The few case reports linking folate deficiency and thiamine deficiency with optic neuropathy have not included OCT, but one patient with folate deficiency did exhibit bilateral temporal disc edema (79-83). These data suggest that selective temporal thinning on OCT in the appropriate clinical context should raise concern for metabolic optic neuropathy.
Optic neuropathy in the setting of vitamin deficiency is often multifactorial. It is likely that the phenomenon referred to as “tobacco-alcohol amblyopia” results from a convergence of vitamin deficiency, the toxic effects of tobacco and alcohol, and genetic predisposition (89). Therefore, clinicians should be extremely cautious about rendering this diagnosis without a thorough investigation for other causes. Patients can present with multiple vitamin deficiencies concurrently, so it is often advisable to test comprehensively for B12, thiamine, folate and copper along with CBC to assess for macrocytic anemia. With the exception of copper, supplementation of the deficient vitamin typically results in excellent visual outcome.
Glaucoma is a common cause of optic neuropathy, with a global prevalence recently estimated at 3.54% in the population aged 40–80 years (90). The optic nerve head typically exhibits deep cupping without pallor until late in the course of disease. In contrast, mitochondrial optic neuropathies typically cause disc pallor within months of symptom onset with a variable amount of cupping. Glaucoma preferentially affects the superior and inferior poles of the optic nerve earliest, with corresponding arcuate visual field defects and sparing of central vision until late in the disease course. Therefore OCT demonstrating RNFL thinning superiorly, inferiorly and nasally is more suggestive of glaucoma, while isolated or more severe temporal thinning with macular GC-IPL thinning early is more characteristic of mitochondrial disease. However, OCT alone is not diagnostic, and the entire clinical picture must be taken into account. As toxic and metabolic optic neuropathies can be easily treated with improvement in visual outcomes, a thorough medication review is recommended and nutritional evaluation should be undertaken in the appropriate clinical context.
Non-arteritic anterior ischemic optic neuropathy (NAION), resulting from microvascular disease, and arteritic anterior ischemic optic neuropathy (AAION), which occurs in the setting of giant cell arteritis (GCA), present with unilateral, acute onset disc edema, vision loss, dyschromatopsia and scotomas that typically respect the horizontal midline. Over the course of 6 to 8 weeks, the disc edema subsides, with the development of disc pallor that is diffuse, or restricted to the superior or inferior aspect of the nerve, with associated RNFL and GC-IPL thinning on OCT. As in glaucoma, the sectoral nature of optic atrophy can be helpful in distinguishing between ischemic optic neuropathies and mitochondrial disorders.
Posterior to the lamina cribrosa, the optic nerve is myelinated and is subject to inflammatory demyelinating diseases including multiple sclerosis and neuromyelitis optica spectrum disorders (NMOSD). Infiltrative inflammatory or malignant processes including sarcoidosis, lymphoproliferative disorders, optic nerve sheath meningioma, and optic nerve gliomas can also involve the orbital or intracanalicular segments of the optic nerve, as can numerous infectious agents including lyme, syphilis and tuberculosis (91). Mass lesions within the orbit can cause a compressive optic neuropathy, and traumatic optic neuropathy results from shear or compressive forces incurred by the optic nerve as it travels through the restrictive space of the optic canal. These diverse etiologies tend to impact the full thickness of the optic nerve to cause decreased visual acuity and color vision, an afferent pupillary defect (APD) (if unilateral), and visual fields with generalized depression. There is sometimes disc edema in acute phases, with later progression to diffuse pallor, with RNFL and GC-IPL thinning on OCT, although sectoral changes have been reported in a subset of cases.
Mass lesions causing compression in the pre-chiasmatic segment of the optic nerve can cause a junctional scotoma, with central vision loss and diffuse disc pallor of the involved side, and a superotemporal scotoma on the contralateral side, while lesions compressing the chiasm classically result in bitemporal hemianopsia, and occasionally cause bilateral temporal pallor of the disc and RNFL thinning on OCT. As discussed above, these findings are seen in a minority of patients with mitochondrial optic neuropathies. Imaging is strongly recommended to rule out mass lesions in the setting of bitemporal or junctional field cuts, particularly those that respect the vertical meridian.
Hereditary, metabolic and toxic optic neuropathies likely result from mitochondrial dysfunction, which disproportionately affects the PMB to cause bilateral central vision loss and dyschromatopsias. Characteristically, OCT shows RNFL thinning that is most pronounced temporally, but this can be delayed in appearance. Concordant ganglion cell/inner plexiform layer (GC-IPL) analysis is particularly useful, as it can show early changes prior to RNFL thinning. Overall, OCT is a valuable tool that can assist in the diagnosis of mitochondrial optic neuropathies.