Abstract: Pediatric neuro-ophthalmology is a subspecialty within neuro-ophthalmology. Pediatric neuro-ophthalmic diseases must be considered separate from their adult counterparts, due to the distinctive nature of the examination, clinical presentations, and management choices. This manuscript will highlight four common pediatric neuro-ophthalmic disorders by describing common clinical presentations, recommended management, and highlighting recent developments. Diseases discussed include pediatric idiopathic intracranial hypertension (IIH), pseudopapilledema, optic neuritis (ON) and optic pathway gliomas (OPG). The demographics, diagnosis and management of common pediatric neuro-ophthalmic disease require a working knowledge of the current research presented herein. Special attention should be placed on the differences between pediatric and adult entities such that children can be appropriately diagnosed and treated.
Pediatric neuro-ophthalmology is a distinct subspecialty from adult neuro-ophthalmology, despite sharing many of the same diagnoses, due to the distinctive nature of the examination, clinical presentations, and management choices. In addition, pediatric neuro-ophthalmic disorders are less commonly seen and less well understood than their adult counterparts. In fact, “Children are not simply little adults” is a frequently quoted mantra of pediatric neuro-ophthalmologists.
The aim of this manuscript is to highlight four common pediatric neuro-ophthalmic disorders by describing common clinical presentations, recommended management, and highlighting recent developments. There has been a recent surge in interest in the management of pediatric neuro-ophthalmic diseases, and therefore a rapid escalation in knowledge has taken place of late. This review will highlight both the standard clinical information required to care for these patients as well as emphasize recent novel research findings.
The estimated overall annual incidence rate of IIH is approximately 0.9 per 100,000 in combined pediatric and adult populations (1-3). However, the demographics of pediatric IIH are different than that of adults. In adult patients, there is a preponderance of female gender and obesity, yet in pre-pubertal pediatric patients, there is an equal distribution of male and female gender, and obesity does not appear to be associated (4-6). Post-pubertal pediatric patients display demographic characteristics more similar to adult patients, with female gender and obesity showing a stronger association (7). Recently, Sheldon and colleagues (8) evaluated anthropometrics of pediatric IIH patients in a multi-center international cohort. Their findings revealed that younger pseudotumor patients (females under the age of 7 years and males under the age of 8.5 years) had normal height and weight, while adolescent patients were more likely to be taller and overweight, and late adolescent children were more typically obese and normal height. The anthropometrics of children with secondary IIH, when due to medication exposure or other systemic factors has not been well evaluated.
In general, the pathophysiology of IIH is related to abnormal cerebrospinal fluid (CSF) dynamics either due to impaired outflow, excessive production, or both. Although adiposity seems to be a predisposing factor for IIH in adults and post-pubertal children, it is not a driving factor in younger children. The pathophysiology in both age groups is still under investigation. One hypothesis involves a unifying mineralocorticoid pathway involving aldosterone, which includes explanations for why elevated vitamin A, obesity and growth hormone are all associated with pseudotumor in children (9). A second theory suggests that altered glucocorticoid metabolism may be the etiology (10). Recent research has revealed that pre-pubertal children with pseudotumor often have low CSF protein concentration, suggesting that increased CSF production may play a role in these children (11). Secondary IIH can be caused by a variety of identifiable causes including medications, medical conditions, structural brain abnormalities, and medication withdrawal (Table 1).
| Medications |
| Tetracycline class of antibiotics |
| Nalidixic acid |
| Vitamin A derivatives |
| Growth hormone |
| Thyroxine |
| Lithium |
| Corticosteroids |
| Corticosteroid withdrawal |
| Medical and genetic conditions |
| Down syndrome |
| Turner syndrome |
| Renal failure |
| Anemia |
| Addison disease |
| Sleep apnea/hypercapnia |
| Vascular abnormalities |
| Cerebral venous sinus thrombosis |
| Jugular vein thrombosis or occlusion |
| Arteriovenous fistulas |
| Remote subarachnoid hemorrhage leading to decreased cerebrospinal fluid absorption |
| Hypercoagulable states |
| Antiphospholipid antibodies |
| Hyperfibrinogenemia |
| Systemic lupus erythematosus |
| Cardiac abnormalities |
| Increased right heart pressure |
| Superior vena cava syndrome |
Clinical symptoms in pediatric pseudotumor patients may be similar to those found in adult patients such as headache, nausea, vomiting, diplopia, transient visual obscuration and pulsatile tinnitus. However, unlike their adult counterparts, pediatric patients can also commonly present with vague symptoms such as irritability or change in behavior, or they may also present without any symptoms in up to 30% of cases (7). Pediatric patients are often referred for evaluation after papilledema or an ocular motility disturbance are noted on routine examination.
Neuro-ophthalmic examination of pediatric patients frequently shows ocular motility abnormalities such as sixth nerve palsies, occurring in 10-40% of patients, or less commonly comitant esotropia or third or fourth cranial nerve palsies (5,7). By far the most common clinical exam finding in these patients is papilledema, which is seen in the majority of cases. Diagnostic criteria for IIH have recently been updated in 2013 (12) in order to include common radiologic characteristics of increased intracranial pressure as well as recent data regarding normal CSF pressure in children. These criteria include (12):
Given the above diagnostic criteria, MRI of the brain combined with MRV is indicated for all cases of suspected IIH syndrome in children. Several recent studies have delved into the MRI findings in children with IIH (13-16). These studies have shown that the most common finding on MRI is optic nerve sheath enlargement (64%) and tortuosity (30–90%) followed by intraocular protrusion of the optic nerve (17–27%), empty sella (26%) and flattening of the posterior globe (45–60%) (13,17). After the MRI/MRV is performed, a lumbar puncture is then indicated both to determine the intracranial pressure and to ensure a normal CSF profile. In addition, recent research has suggested a role for other ancillary tests such as optical coherence tomography (OCT) of the optic nerve and macula. OCT has been suggested as a surrogate for subsequent lumbar puncture in cases where response to treatment is unclear. For example, one study found that optic atrophy and photoreceptor dropout were highly correlated with vision loss and progression of disease (18). A similar study indicated that increased retinal nerve fiber layer (rNFL) and macular thickness can be seen by OCT and used as a tool for assessing and monitoring disease (19). Despite these findings, the optimal OCT parameters for use in monitoring disease progression are still debatable and may include the amount of inward deflection of Bruch membrane (20,21), the macular ganglion cell layer inner plexiform layer (20), optic nerve volume (22), or the retinal nerve fiber layer.
Once the diagnosis of IIH is established, treatment is based on the severity of the individual patient’s presentation. Treatment goals include alleviation of symptoms such as headache, diplopia, and visual changes as well as prevention of permanent vision loss from papilledema. Typically, asymptomatic patients with normal visual acuity and visual fields can be managed conservatively with observation or low dose acetazolamide. In addition, in cases of secondary IIH, the predisposing factor must also be addressed. In cases where there are bothersome symptoms or mild visual compromise, medical management with acetazolamide 15–25 mg/kg/day divided into two or three dosages, increasing as necessary up to 100 mg/kg/day (up to 2 grams per day) is typically the first line therapy. If acetazolamide is not tolerated due to side effects, then alternative medications such as topiramate and furosemide can be considered. Medications can be tapered once the papilledema resolves and the visual field normalizes. Frequent monitoring of visual acuity, visual field and optic nerve appearance is crucial to avoid untreated worsening of disease. In fulminant or severe acute cases, occasionally surgical intervention is required. As a temporizing measure in some patients, intravenous steroids are utilized in attempt to minimize severe visual loss. Surgical intervention may include optic nerve sheath fenestration or CSF shunting depending on the patient’s symptomatology as well as the preference of the treating center. In all cases, attention should be given to the patient’s body mass index and weight loss encouraged in cases of obesity or overweight status.
The Idiopathic Intracranial Hypertension Treatment Trial was the first randomized controlled trial for treatment of IIH in adults (23). Although no children were enrolled in this study, the data gleaned from it may have important implications for pediatric patients. For example, this study determined that worse visual acuity or papilledema at presentation was associated with worse outcomes—this could possibly be extrapolated to children and may encourage clinicians to treat these patients more aggressively at disease onset. In addition, on the horizon for the management of IIH are more studies evaluating the use of various OCT parameters to monitor and diagnose IIH, potentially avoiding invasive testing such as lumbar puncture. Lastly, the pathophysiology of IIH must still be elucidated in order to create relevant treatments especially in those patients with the most severe presentations.
Pseudopapilledema in children is a common diagnosis on the differential diagnosis of possible papilledema. A common cause of pseudopapilledema is ODD, which are benign congenital deposits in the optic nerve head. ODD are most commonly buried in the substance of the optic nerve head, and therefore not clearly visible in pediatric patients. Pseudopapilledema may be mistaken for true papilledema in up to 76% of cases (24), leading to unnecessary, expensive and invasive testing. Therefore, it is crucial for ophthalmologists to be able to differentiate true papilledema from pseudopapilledema. The prevalence of ODD in children is estimated at approximately 0.4%; however, in adults estimates are higher and range from 0.5% to 2.4%, probably because they are easier to detect in adults (25-27). ODD prevalence also varies by race, with a higher prevalence in white patients (28). ODD tend to occur more commonly in children with a family history of ODD and may be inherited as part of genetic syndromes such as retinitis pigmentosa, pseudoxanthoma elasticum (29,30), and Alagille syndrome (31).
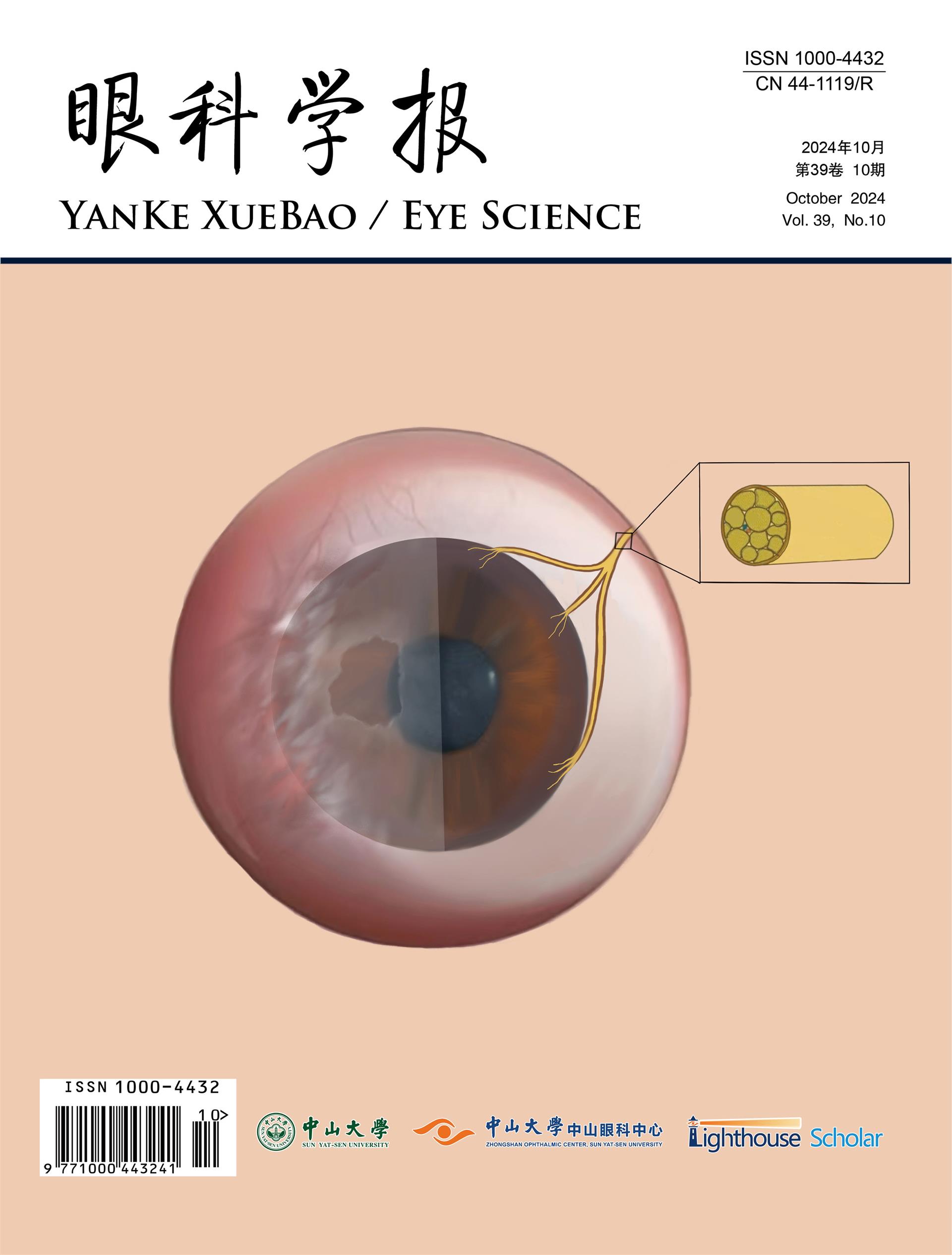
ODD appear different in pediatric patients compared to adults. In children, ODD are frequently buried within the substance of the optic nerve (Figure 2) while in adults, they are situated on the surface of the disk and are often calcified. Due to these differences in appearance, ODD in children is more difficult to visualize and diagnose.
The exact cause of ODD is not known but there are several theories proposed to explain their presence. The two most commonly accepted theories are that the drusen are caused by (I) a disturbance of axonal metabolism with slow axoplasmic flow (32,33) or (II) congenitally small or dysplastic disks with a propensity for altered axoplasmic flow and subsequent drusen formation (34,35). The unifying feature of many hypotheses is that patients with drusen are anatomically predisposed due to a small scleral canal (or disturbed axonal flow in cases of normal-sized scleral canals) (25).
ODD in children are almost always found incidentally on routine eye examination, as the majority of cases are asymptomatic. Previous series have revealed that approximately half of children evaluated for ODD present with symptoms of increased ICP such as headache, nausea, or vomiting, while the other half of patients are diagnosed as part of an ophthalmic examination for unrelated issues (36). Rarely, children can present with symptoms from ODD that can include transient visual obscuration or visual field defects (37-39). Visual field defects occur with increasing frequency as the age of the patient increases, as visual field defects are more commonly seen with surface drusen (40,41). The incidence of visual field defects in young (<10 years) patients with ODD is estimated at 11% compared to 51% in older children (28). The most common visual field defect seen in these cases are nasal steps, arcuate defects, enlarged blind spots, and generalized constriction (38). Aside from visual field defects, possible examination findings in children with ODD include anomalous retinal vasculature, disk hemorrhage, relative afferent pupillary defect and very rarely choroidal neovascular membrane.
The difficulty in the management of children with pseudopapilledema is differentiating their presentation from true papilledema. For this reason, there are many studies evaluating various diagnostic tests in attempt to determine the most accurate way to differentiate these diagnoses without requiring expensive or invasive tests such as MRI or lumbar puncture. Ancillary tests vary in their level of accuracy in differentiating true edema from pseudopapilledema in children (Table 2).
| Diagnostic test | Buried (%) | Surface (%) |
|---|---|---|
| Fundus autofluorescence | 56 | 100 |
| Fluorescein angiography | 95 | 100 |
| Optical coherence tomography | SD: 82 | SD: 100 |
| EDI: 76 | EDI: 100 | |
| RNFL: 79 | RNFL: 67 | |
| B-scan ultrasonography | 84 | 100 |
*, data from Chang
B-scan ultrasonography is an inexpensive and non-invasive modality that is very useful in diagnosing ODD in adults (43,44). The diagnosis is based upon a hyperechoic mass within the substance of the optic nerve associated with posterior shadowing. However, this finding is only seen when ODD are calcified, and therefore has been found to have a sensitivity of approximately 48% in adult populations (43). The sensitivity in children is likely lower due to the lack of calcifications; however, in a recent pediatric study, B-scan was found to have a 74% accuracy in differentiating true papilledema from pseudopapilledema (42).
ODD tend to autofluoresce on pre-injection control photography and scanning laser ophthalmoscopy (28). Similar to B-scan, autofluorescence has a higher utility in surface, calcified ODD as the autofluorescent nature of buried drusen is obscured by the overlying optic nerve tissue. In children with surface drusen confirmed by B-scan, autofluorescence is 94% sensitive (45). However, in studies that did not require B-scan confirmation (and are therefore likely to include more children with buried drusen), the detection of buried drusen by autofluorescence ranged from 27–56% (42,43).
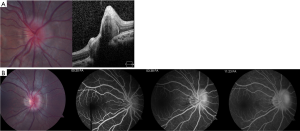
FA is a useful but more invasive test that is often helpful in differentiating true from pseudopapilledema. ODD demonstrate several common characteristics on FA, including optic disk staining, delayed filling of the peripapillary choriocapillaris, lack of early or late disk leakage, and staining of the ODD (Figure 2) (46). FA has been shown to have the highest accuracy (97%) of all tested modalities in differentiating ODD from true papilledema in a recent study of pediatric patients (42). Limitations of FA include the need for intravenous injection which may be obviated by the use of oral fluorescein (47).
There has been a surge of interest in the use of OCT to diagnose ODD and differentiate it from true papilledema. Modalities of OCT that have been suggested include retinal nerve fiber layer analysis and direct visualization using enhanced depth imaging (EDI) or swept source OCT. On OCT, ODD appear as focal masses with a hyporeflective core and hyperreflective border (Figure 2). There has been some controversy as to whether OCT is able to accurately differentiate ODD from true papilledema. Recently, the Optic Disc Drusen Studies Consortium has published recommendations for the diagnosis of ODD using OCT (48). Their recommendations include a standardized protocol for pre-scanning and acquisition of scans, and specify specific parameters for dense optic nerve scans, radial optic nerve scans, peripapillary scans, and macula scans (48). The purpose of these recommendations is to establish reliable and consistent diagnosis of ODD both for researchers and clinicians. Although their recommendations are crucial, they may not be directly applicable to pediatric patients as their standardization steps were based on adult patients with less than 50% harboring buried drusen (vs. surface drusen). Their findings suggest that on EDI OCT, ODD are always located above the lamina cribosa, have a signal-poor core and are often seen with a hyperreflective margin (48).
Studies that have focused specifically on pediatric patients have shown conflicting results as to whether OCT is useful in differentiating ODD from papilledema. In one series, EDI OCT detected 17/25 cases of buried ODD (49). In a more recent study of EDI-OCT in pediatric patients, the accuracy was 67% for differentiating papilledema from ODD (42).
Few studies have compared various modalities for the diagnosis of ODD (42,43). Recently, Chang et al. published a study comparing B-scan, autofluorescence, FA, and OCT in the differentiation of pediatric pseudopapilledema and true papilledema cases (42). The findings of this study (Table 2) revealed that FA had the highest diagnostic accuracy in differentiation the two entities (97%), followed by ultrasonography (74%), OCT (67–71%), fundus photography (66%), and autofluorescence (62%).
Despite ODD being a benign entity, there are rare complications that may cause loss of vision in some patients. As discussed above, visual field defects occur in some children with ODD and increase with age. ODD may also be associated with vascular complications such as hemorrhages (50-52), choroidal neovascular membrane (53), vascular occlusions and even ischemic optic neuropathy (54). These vascular complications are rare but have been reported.
Given the risk of visual field defects and other rare complications, it is recommended that children with ODD undergo at least annual ophthalmic examinations and visual field testing. In cases where visual field loss is detected, one can consider ocular hypotensive drops although no studies have been performed to support this recommendation (28). Clinicians must keep in mind that alpha agonists are contraindicated in young children due to the risk of respiratory depression.
Several groups have also identified changes in the Bruch’s membrane opening on OCT as an aid to differentiating true papilledema from pseudopapilledema (18,55,56). This area is still one of active investigation but appears promising in that the horizontal diameter of the Bruch membrane opening appears enlarged in patients with true papilledema compared to pseudopapilledema patients. In addition, future studies may evaluate neuroprotective agents for use in ODD patients who appear at risk for visual field loss.
ON is a clinical diagnosis defined as an idiopathic, demyelinating inflammatory condition of the optic nerve. Non-idiopathic cases, such as those caused by autoimmune and inflammatory disorders like sarcoidosis or systemic lupus erythematosus are often grouped into the same diagnostic categories. Pediatric ON may occur in isolation or as part of an inflammatory central nervous system disorder such as acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS) or neuromyelitis optica (NMO), and it accounts for approximately 25% of acute demyelinating events in children (57). The overall incidence of pediatric ON has been estimated at 0.2 per 100,000 in Canada (57). In pre-pubertal children, there does not appear to be a gender predilection; however, in post-pubertal children, there is a preponderance of female patients (58,59).
The typical presentation of pediatric ON includes decreased visual acuity, dyschromatopsia, and visual field defects. Unilateral or bilateral involvement may occur, and bilateral involvement may be simultaneous (within 2 weeks) or sequential (2–12 weeks). Bilateral involvement is more common in younger children (72% <10 years) than in older children (30%) (60). Pain with eye movements or headache is a presenting symptom in 30–77% of patients (61-64). The most common visual field defects in those patients who are able to perform automated visual field testing are central or cecocentral scotoma (61,65).
Visual acuity at presentation may be relatively normal with approximately 20% of children presenting with visual acuity better than 20/40. More commonly, it is severely affected, with 60% of children presenting with VA worse than 20/200 (64,66). In case series that include only pre-pubertal children, visual acuity at presentation is worse, with approximately 90% of children presenting with VA worse than 20/200 (67). Common examination features include optic disk edema (40–70%) (61,64,68), relative afferent pupillary defect, or optic nerve pallor if recurrent or late stage. Long-term outcomes after ON in children are typically favorable. Most (58–97%) children recover visual acuity better than 20/40 (58,64,66,69), and in the most recent study of children requiring a minimum of one-year follow up, 89% of patients recovered vision better than 20/40 at 1 year (63). Structural rNFL outcomes vary depending on the underlying etiology, with ADEM patients faring the worst (70).
There are several important differences between pediatric and adult patients with ON. With regard to presentation, adult patients more frequently present with pain on eye movements than children (90% vs. 53%) (68). In addition, the majority of adult patients present with unilateral retrobulbar involvement, in contrast to pediatric patients who typically present with bilateral optic nerve edema (one-third vs. 40–70%) (61,66,71). Presenting VA is typically better in adults than children, with 36% of adults in the Optic Neuritis Treatment Trial presenting with VA worse than 20/200 compared to 90% of pre-pubertal ON patients (67,72).
Although ON is a clinical diagnosis, it requires a work-up to rule out other causes of optic disk edema in children (Table 3).
| Demyelinating optic neuritis |
| Acute disseminated encephalomyelitis (ADEM) |
| Neuromyelitis optica spectrum disorder(NMOSD) |
| Multiple sclerosis (MS) |
| Isolated optic neuritis (post-viral, post-vaccine) |
| Systemic rheumatologic condition |
| Systemic lupus erythematosus |
| Sarcoidosis |
| Small vessel vasculitis |
| Chronic relapsing inflammatory optic neuropathy |
| Genetic optic atrophy e.g., Leber hereditary optic neuropathy |
| Infectious etiologies |
| Syphilis |
| Lyme |
| Bartonella |
| Toxoplasma |
| Orbital cellulitis |
| Sinusitis |
| Infiltrative disease |
| Leukemia |
| Lymphoma |
| Papilledema |
| Pseudopapilledema |
| Non-accidental trauma |
| Optic nerve or nerve sheath tumor e.g., meningioma, glioma |
Typical investigations include magnetic resonance imaging (MRI) of the brain and orbits as well as a lumbar puncture with opening pressure, CSF analysis for protein, glucose, cells, oligoclonal bands, IgG index and cultures. MRI testing typically reveals optic nerve thickening on T1-weighted images and bright signal on T2-weighted images. Of note, lumbar puncture opening pressure may be elevated in cases of ON, occasionally causing difficulty in differentiating bilateral ON from IIH (73,74). Laboratory testing can be guided by the presentation but typically includes testing for biomarkers of sarcoidosis, tuberculosis, syphilis, Lyme disease, Bartonella henselae infection, and systemic lupus erythematosus. If there is suspicion for NMO, an MRI of the spine as well as aquaporin-4 testing can be performed.
ON in children may be an isolated monophasic event or it may be post-infectious, inflammatory or part of a systemic demyelinating disorder. In younger children, ON is more likely to occur as a post-viral or post-vaccination syndrome or in association with ADEM, while older children tend to present more commonly as an initial manifestation of a systemic demyelinating disease such as MS or NMO.
Isolated ON is more common in children than adults and is often associated with an antecedent viral infection, vaccination or non-viral infection such as Lyme disease or Mycoplasma pneumonia (59,75). Recurrent ON in children without anti-aquaporin-4 antibody positivity may be associated with positive oligoclonal bands or anti-MOG. Recurrent ON in isolation is uncommon, occurring in 5% of Canadian pediatric ON patients (57). Multiple recurrences of ON that are associated with objective loss of visual function, negative anti-NMO status, and are responsive to steroids, with relapse upon their withdrawal may be known as chronic relapsing inflammatory optic neuropathy (CRION).
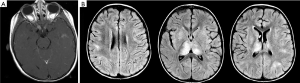
ADEM is a monophasic (typically) multifocal acute demyelinating disease that presents mostly in prepubertal children typically following a viral illness or vaccination (76). ADEM is characterized by encephalopathy, fever, and meningeal signs as well as MRI findings of T2 and flair hyperintense large white matter lesions characterized by uniformity in time of onset (Figure 3) (77). ADEM is typically differentiated from MS by its lack of dissemination in time and lack of recurrence although multi-phasic ADEM does rarely occur (76).
MS in children is diagnosed similarly to adults, using the McDonald criteria. However, when children present with ON prior to the age of 12 years, the positive predictive value of the McDonald criteria is lower (76). ON is the presenting feature of MS in 10–20% of pediatric MS cases (78). The risk of developing MS after an initial episode of ON is highly dependent on the age at presentation, MRI findings, and CSF findings (OCB positivity) Although it is estimated that 13–36% of pediatric patients presenting with ON will eventually be diagnosed with MS (62,66,71), this risk is highly dependent on the age at presentation as well as the presence or absence of white matter lesions on the initial brain MRI (64,71). For every 1-year increase in age, it has been estimated that there is a 32% increased risk of MS. In addition, a recent meta-analysis revealed that the presence of one white matter lesion on brain MRI increased the odds ratio of an eventual diagnosis of MS to 28.0 (60). In a European study, the hazard ratio for MS was 1.08/year of age (79). However, the risk of MS also depends upon racial and geographic features (59).
NMO is traditionally characterized by ON and transverse myelitis with characteristic MRI findings of longitudinally extensive optic nerve and transverse myelitis lesions. A recent revision of the international consensus diagnostic criteria for NMO spectrum disorder was categorized by anti-aquaporin-4 antibody status, and included the presence of hyperintense optic nerve lesions on T2-weighted imaging (or T1-weighted gadolinium enhancing lesions) extending more than 50% of the length of the optic nerve and transverse myelitis lesions spanning at least 3 contiguous spinal segments (80). The consensus group noted that spinal lesions are less diagnostic in children as they can also occur in ADEM and pediatric MS. ON associated with NMO in children is frequently associated with poorer visual outcomes with the median time from onset of NMO to severe vision loss of <20/200 estimated to be only 1.3 years (81).
The decision to treat pediatric ON varies amongst physicians and is often dependent upon the age of the patient, systemic comorbidities laterality, MRI findings, and severity of vision loss. Acute treatment typically includes the administration of corticosteroids, commonly intravenous methylprednisolone for a course of 3–5 days (20–30 mg/kg/day up to 1 g/day) extrapolated from the adult ON treatment trial (82). Whether a longer (>2 weeks) vs. shorter (<2 weeks) oral steroid taper affects outcome is still debatable (83). Since there are no prospective randomized trials, it is unclear what benefit corticosteroid treatment in general may provide in the long-term. In cases of severe steroid-resistant ON, IVIG and plasma exchange can be considered. If a diagnosis of MS or NMO is made, systemic disease-modifying therapy is recommended.
Several ancillary tests are still under investigation for their utility in the diagnosis and management of pediatric ON. For example, OCT measurements of the rNFL and ganglion cell layer may have a role in long-term follow up for patients with and without MS (84-86). Structure-function studies will help to determine the patient-related nature of these findings. Additionally, anti-myelin oligodendrocyte glycoprotein antibody (anti-MOG), an antibody that is prevalent in younger ON patients with encephalopathy, may be a useful adjunct test in cases where the presentation is not typical for MS, ADEM or NMO (87). Preliminary studies have shown that anti-MOG positivity may be associated with better outcomes compared to other demyelinating diseases in children (88,89). The relationship between anti-MOG antibody and ADEM as well as recurrent ON in children remains to be examined.
The lack of prospective data on the natural history and treatment of children with ON limits our ability to practice evidence-based management in these cases. A recently launched collaborative study between the Pediatric Eye Disease Investigator Group (PEDIG) and the Neuro Ophthalmology Research Disease Investigator Consortium (NORDIC) aims to enroll 100 children with acute ON amongst 45 sites over a period of 2 years. This prospective data collection study will determine the group’s ability to enroll children for a potential future prospective treatment study as well as to estimate visual acuity outcomes at 6 months, and numerous secondary outcomes such as quality of life metrics, OCT, low contrast acuity, MRI findings and NMO antibody status.
OPG are low-grade glial neoplasms composed of astrocytes involving the visual pathway anywhere along the path from the optic nerves to the optic radiations. OPGs are typically diagnosed based on their characteristic appearance on neuroimaging (see below) but have also been examined pathologically. The histological appearance of these tumors is benign and includes Type 1 astrocytes with similar molecular genetic makeup to other low-grade astrocytomas in the CNS (90-93). Recent debate in the literature as to whether these tumors should be considered “benign hamartomas” instead of “pilocytic astrocytomas” has been discussed; most experts agree that the identical molecular make-up of the tumors to other CNS astrocytomas as well as their immunohistochemical profile determines that the most accurate name for the lesions is pilocytic astrocytoma (93).
OPGs are relatively common in children with neurofibromatosis-1 (NF1) occurring in 15–20% of them, but in the general population are more rare, accounting for approximately 1% of all intracranial tumors and 3–5% of pediatric brain tumors (93,94). The majority (70%) of OPGs present in the first decade of life (median age 6.5 years) although rarely elderly patients have been reported to present with a seemingly new onset OPG (95-97).
The clinical presentation of OPGs is dependent upon the age at presentation, laterality and location of the lesion. Unilateral anterior optic nerve gliomas are typically associated with decreased visual acuity, dyschromatopsia, and visual field defects. Examination findings may include a relative afferent pupillary defect, proptosis, strabismus, or optic disk edema. More posterior optic nerve tumors may present more insidiously with optic atrophy and less external findings. OPGs of the chiasm are frequently diagnosed later than anterior tumors since there are less external features, and the visual loss is more peripheral and has a slower onset. Visual loss is bilateral and typically associated with bitemporal hemianopia. Hemianopic defects in these cases are typically asymmetric and many patients present with a junctional scotoma, with central vision loss in one eye and a temporal field defect in the fellow eye. Examination findings include optic atrophy and rarely proptosis if the tumor extends anteriorly into one or both optic nerves. Other possible presenting examination findings include sensory strabismus, asymmetric nystagmus, and hypothalamic dysfunction. Nystagmus associated with chiasmal gliomas can occasionally appear similarly to spasmus nutans with an asymmetric horizontal shimmering waveform.
Anterior OPGs are typically diagnosed based on neuroimaging characteristics which include fusiform enlargement and kinking of the optic nerve. OPGs are iso- or hypointense on T1-weighted imaging with enhancement after IV contrast administration, and are hyperintense on T2-weighted images. These imaging characteristics are consistent across all possible locations of OPGs. In diagnosing OPGs, it is important to look for other MRI findings seen in NF1 if the patient’s NF1-status is not known. These findings may include focal areas of abnormal signal intensity (previously known as “unidentified bright objects”) which are thought to be the result of neural dysplasia or dysmyelination with associated focal edema (98). In addition, physical examination should include an assessment for other findings seen in patients with NF1 such as café-au-lait spots, neurofibromas, axillary or inguinal freckling, Lisch nodules, and bone changes such as bowing of the long bones, and a genetics consultation if indicated.
Once diagnosed, the natural history of OPGs is typically characterized by slow growth, minimal effect on visual acuity, and exceedingly rare potential for malignant transformation (93). Interestingly, OPGs that are isolated and not associated with NF1 seem to have a more aggressive course with worse visual outcomes. Children with sporadic OPGs have an approximate rate of vision loss of 70% while patients with NF1-related OPGs have a lower rate of vision loss of approximately 25% (99-105). Isolated optic nerve lesions are known to be associated with the presence of some cells in the chiasm, even if not visible radiographically (106). However, the risk of radiographically visible progression of an isolated optic nerve tumor to “invade” the chiasm is rare, occurring in 7 of 35 patients followed in a national clinical trial (107). Negative prognostic factors for visual acuity that have been posited but not confirmed in a large randomized trial include female gender, earlier age of presentation (108), and more anterior location of the tumor (99,109-113). Chiasmal gliomas are typically less aggressive than optic nerve tumors, but may be associated with more systemic complications such as hypopituitarism. The most posterior lesions (optic tract and radiations) appear to be the least aggressive lesions in terms of growth (99,111-113).
The management of OPGs is still controversial as their natural history is not completely agreed upon, and there are case reports of spontaneous improvement (114). However, treatment is indicated to reduce visual morbidity in children who are at risk. Furthermore, in patients with NF1, it has been shown that vision loss, endocrinopathy, impaired social interactions, and difficulties with activities of daily living are all associated with OPGs (115). Recently, there has been a paradigm shift away from tumor size increase as an indication for treatment, with more emphasis placed on visual acuity loss (or its biomarkers) as the primary indication for treatment (115). Although there is no clear consensus for treatment indications, most clinicians agree that the best indication for treatment is significant vision loss (a change of 0.2 logMAR or progression of visual field defects) (115). Other less common scenarios that may indicate the need for treatment include abnormal vision in the fellow eye of an already blind eye, suspected vision loss in the absence of an obtainable visual acuity, or visual acuity at or near a functional threshold (100,115). Tumor growth without a change in visual acuity does not indicate a need for treatment; however, progression on MRI along with changes to the physical examination may be an indication for closer surveillance or treatment.
The first line treatment for OPGs is typically chemotherapy, as radiation carries a risk of secondary malignancy, moyamoya-like disease and neurocognitive deterioration in young children. Common chemotherapeutic options include vincristine, vinblastine, carboplatin, temozolomide, and most recently bevacizumab (116). Surgery is reserved for extreme cases, such as physical deformity or encroachment onto vital CNS structures. Despite the use of chemotherapy in children with progressive vision loss, some children continue to deteriorate with 28% of children deteriorating by at least 2 logMAR lines after the completion of chemotherapy in one study (110).
In patients with NF1 without a known OPG, current recommendations include a yearly comprehensive ophthalmologic examination assessing VA, visual fields, pupillary testing, eye movements, and fundus examination annually until the age of 8 years and then every other year until 18 years of age (100). In those NF1 patients with known associated OPGs, the current recommendation for surveillance by ophthalmology and MRI is every 3 months for the first year and then every 6 months for 2 years and after the age of 8 years, and then annually until 18 years of age (MRI can be less frequent after the first 5 years if there is no progression) (115). Visual acuity testing should be performed when possible using age-appropriate optotypes. However, given the variable nature of visual acuity testing in children, investigators have sought other biomarkers for vision loss. Modalities that have received recent attention include visual evoked potential (117-119), OCT (120-124), diffusion tensor imaging (125,126), and volumetric MRI (127). The two most promising of these modalities appear to be OCT for anterior lesions and diffusion tensor imaging for lesions of the optic tract and radiations.
OCT has been demonstrated to reveal reduced rNFL in children with OPGs, with an apparent threshold near 80 μm, below which patients are more likely to have abnormal visual function (VA or visual field). This threshold may be potentially useful in children who cannot participate with VA testing. Longitudinal studies have also reported the utility of OCT in diagnosing vision loss from OPGs, with a 10% decrease in rNFL thickness indicating vision loss with 70% sensitivity. The overall role of OCT in the management of patients with OPG is still under investigation. Diffusion tensor imaging is an MRI technique that is useful in diagnosing white matter damage. This technique has been shown in a recent study to correlate with vision loss in patients with OPGs of the optic radiations (115). In a small study, this technique was also useful in predicting visual acuity change one year after imaging (126).
A multicenter NF1-OPG study examining the natural history of OPGs has recently launched. This study will help to define visual acuity outcomes as well as biomarkers that may be useful in future treatment trials. In addition, a randomized treatment trial comparing vinblastine and bevacizumab to vinblastine alone has also recently been launched. Subsequent treatment trials comparing various modalities are likely to follow suit. Our understanding of the molecular alterations that underlie OPGs has led to the establishment of newer targeted treatments such as mitogen-activated protein kinase (MEK) inhibitors. Future trials will delineate whether MEK inhibitors, as well as other therapies such as mammalian target of rapamycin complex (mTOR) inhibitors and farnesyl transferase inhibitors are effective the treatment of these tumors (128).