Background:Diabetic retinopathy (DR) urgently needs novel and effective therapeutic targets. Integrated analyses of plasma proteomic and genetic markers can clarify the causal relevance of proteins and discover novel targets for diseases, but no systematic screening for DR has been performed.
Methods: Summary statistics of plasma protein quantitative trait loci (pQTL) were derived from two extensive genome-wide analysis study (GWAS) datasets and one systematic review, with over 100 thousand participants covering thousands of plasma proteins. DR data were sourced from the largest FinnGen study, comprising 10,413 DR cases and 308,633 European controls. Genetic instrumental variables were identified using multiple filters. In the two-sample MR analysis, Wald ratio and inverse variance-weighted (IVW) MR were utilized to investigate the
causality of plasma proteins with DR. Bidirectional MR, Bayesian Co-localization, and phenotype scanning were employed to test for potential reverse causality and confounding factors in the main MR analyses. By systemically searching druggable gene lists, the ChEMBL database, DrugBank, and Gene Ontology database, the druggability and relevant functional pathways of the identified proteins were systematically evaluated.
Results: Genetically predicted levels of 24 proteins were significantly associated with DR risk at a false discovery rate <0.05 including 11 with positive associations and 13 with negative associations. For each standard deviation increase in plasm protein levels, the odds ratios (ORs) for DR varied from 0.51 (95% CI: 0.36-0.73; P=2.22×10-5) for tubulin polymerization-promoting protein family member 3 (TPPP3) to 2.02 (95% CI: 1.44-2.83; P=5.01×10-5) for olfactomedin like 3 (OLFML3). Bidirectional MR indicated there was no reverse causality that interfered with the results of the main MR analyses. Four proteins exhibited strong co-localization evidence (PH4 ≥0.8): cytoplasmic tRNA synthetase (WARS), acrosin binding protein(ACRBP), and intercellular adhesion molecule 1 (ICAM1) were negatively associated with DR risk, while neurogenic locus notch homolog protein 2 (NOTCH2) showed a positive association. No confounding factors were detected between pQTLs and DR according to the phenotypic scan. Drugability assessments highlighted 6 proteins already in drug development endeavor and 18 novel drug targets, with metalloproteinase inhibitor 3 (TIMP) currently in phase I clinical trials for DR. GO analysis identified 18 of 24 plasma proteins enriching 22 pathways related to cell differentiation and proliferation regulation.
Conclusions:Twenty-four promising drug targets for DR were identified, including four plasma proteins with particular co-localization evidence. These findings offer new insights into DR's etiology and therapeutic targeting, exemplifying the value of genomic and proteomic data in drug target discovery.
Background:Diabetic retinopathy (DR) urgently needs novel and effective therapeutic targets. Integrated analyses of plasma proteomic and genetic markers can clarify the causal relevance of proteins and discover novel targets for diseases, but no systematic screening for DR has been performed.
Methods: Summary statistics of plasma protein quantitative trait loci (pQTL) were derived from two extensive genome-wide analysis study (GWAS) datasets and one systematic review, with over 100 thousand participants covering thousands of plasma proteins. DR data were sourced from the largest FinnGen study, comprising 10,413 DR cases and 308,633 European controls. Genetic instrumental variables were identified using multiple filters. In the two-sample MR analysis, Wald ratio and inverse variance-weighted (IVW) MR were utilized to investigate the
causality of plasma proteins with DR. Bidirectional MR, Bayesian Co-localization, and phenotype scanning were employed to test for potential reverse causality and confounding factors in the main MR analyses. By systemically searching druggable gene lists, the ChEMBL database, DrugBank, and Gene Ontology database, the druggability and relevant functional pathways of the identified proteins were systematically evaluated.
Results: Genetically predicted levels of 24 proteins were significantly associated with DR risk at a false discovery rate <0.05 including 11 with positive associations and 13 with negative associations. For each standard deviation increase in plasm protein levels, the odds ratios (ORs) for DR varied from 0.51 (95% CI: 0.36-0.73; P=2.22×10-5) for tubulin polymerization-promoting protein family member 3 (TPPP3) to 2.02 (95% CI: 1.44-2.83; P=5.01×10-5) for olfactomedin like 3 (OLFML3). Bidirectional MR indicated there was no reverse causality that interfered with the results of the main MR analyses. Four proteins exhibited strong co-localization evidence (PH4 ≥0.8): cytoplasmic tRNA synthetase (WARS), acrosin binding protein(ACRBP), and intercellular adhesion molecule 1 (ICAM1) were negatively associated with DR risk, while neurogenic locus notch homolog protein 2 (NOTCH2) showed a positive association. No confounding factors were detected between pQTLs and DR according to the phenotypic scan. Drugability assessments highlighted 6 proteins already in drug development endeavor and 18 novel drug targets, with metalloproteinase inhibitor 3 (TIMP) currently in phase I clinical trials for DR. GO analysis identified 18 of 24 plasma proteins enriching 22 pathways related to cell differentiation and proliferation regulation.
Conclusions:Twenty-four promising drug targets for DR were identified, including four plasma proteins with particular co-localization evidence. These findings offer new insights into DR's etiology and therapeutic targeting, exemplifying the value of genomic and proteomic data in drug target discovery.
INTRODUCTION
Diabetic retinopathy (DR) is the leading cause of blindness in the working-age population, affecting 103.12 million individuals globally. [1] Although the VEGF pathway plays a pivotal role in DR pathogenesis, responses to anti-VEGF treatments vary significantly among patients.[2] Even with close monitoring and frequent injections, achieving desired outcomes remains challenging, with up to 40% cases of diabetic macular edema (DME) experience unresolved edema.[3] This underscores the urgent need for new DR therapeutic targets.
The plasma proteome, reflecting human health and disease, offers crucial insights into an individual's molecular profile.[4] High-throughput proteomics techniques have revolutionized the discovery of disease-related proteins (e.g., dementia, heart failure, cancer) at an unprecedented pace.[5-6] Moreover, the extensive coverage and ready accessibility of plasma proteins make them promising targets for pharmaceutical interventions, with 75% of 2017 FDA-approved drugs targeting human proteins.[7]
Mendelian randomization (MR) minimizes confounding and enables causal inference for exposure (plasma proteins)-outcome (DR) relationships. Genome-wide association studies (GWAS) have identified over 18,000 protein quantitative trait loci (pQTLs), forming a foundation for MR analysis.[8-14] Based on these, proteome-wide MR (PW-MR) has recently yielded significant insights into the etiology of various conditions, including stroke, psychiatric disorders, and breast cancer.[15-17] Moreover, PW-MR offers exceptional prospects for identifying and prioritizing drug targets compared to other approaches.[18] However, no large-scale PW-MR study for DR is currently available. Therefore, this study aimed to identify plasma proteins that could serve as potential therapeutic targets for DR by integrating human plasma proteomic and genomic data, and systematically assessing the druggability of potential DR therapeutic target proteins.
MATERIALS AND METHODS
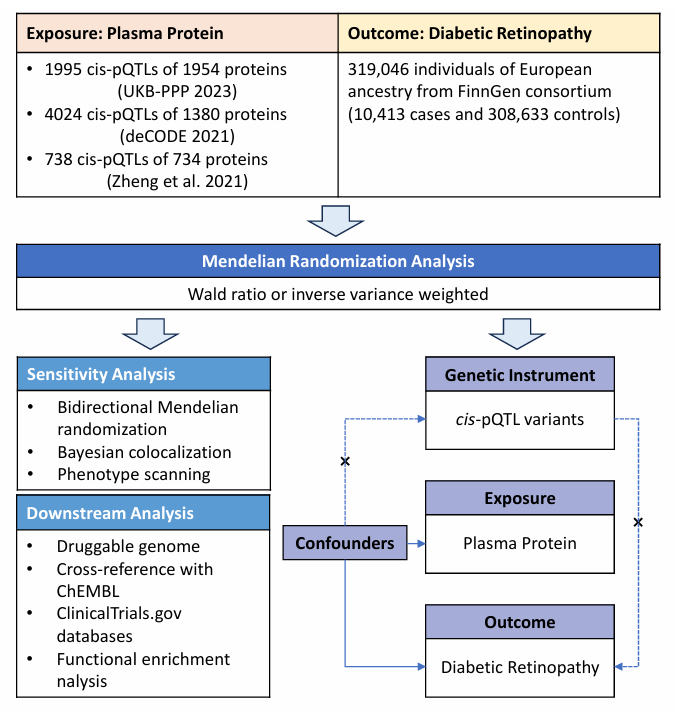
Figure 1 presents the study design. In brief, pQTL data from seven large-scale proteomic studies were utilized to examine their correlation with DR using a two-stage PW-MR methodology. Bayesian co-localization was subsequently applied to validate the causal linkage between proteins and DR. A thorough assessment of the drugability of the identified proteins followed, with a specific emphasis on their potential as therapeutic targets.
Figure 1 A flow chart of the study design and a schematic illustration of cis-MR
Study Exposures: pQTL
Instrument variable (IV) for plasma proteins, specifically cis-pQTLs, were sourced from two proteomic GWASs and a PW-MR study. These cis-pQTLs were gathered from two large-scale GWAS datasets: the UK Biobank Pharma Protein Project (UKB-PPP)[19] and the deCODE Health study.[9] We also obtained data from Zheng et al.,[20] which encompassed 734 plasma proteins with cis-pQTLs. Our selection criteria for inclusion included: (Ⅰ) genome-wide significance (P < 5 × 10-8); (Ⅱ) independence (linkage disequilibrium [LD] clump R2 < 0.001); and (Ⅲ) proximity as cis-pQTLs within 1 Mb of the respective protein-encoding gene.
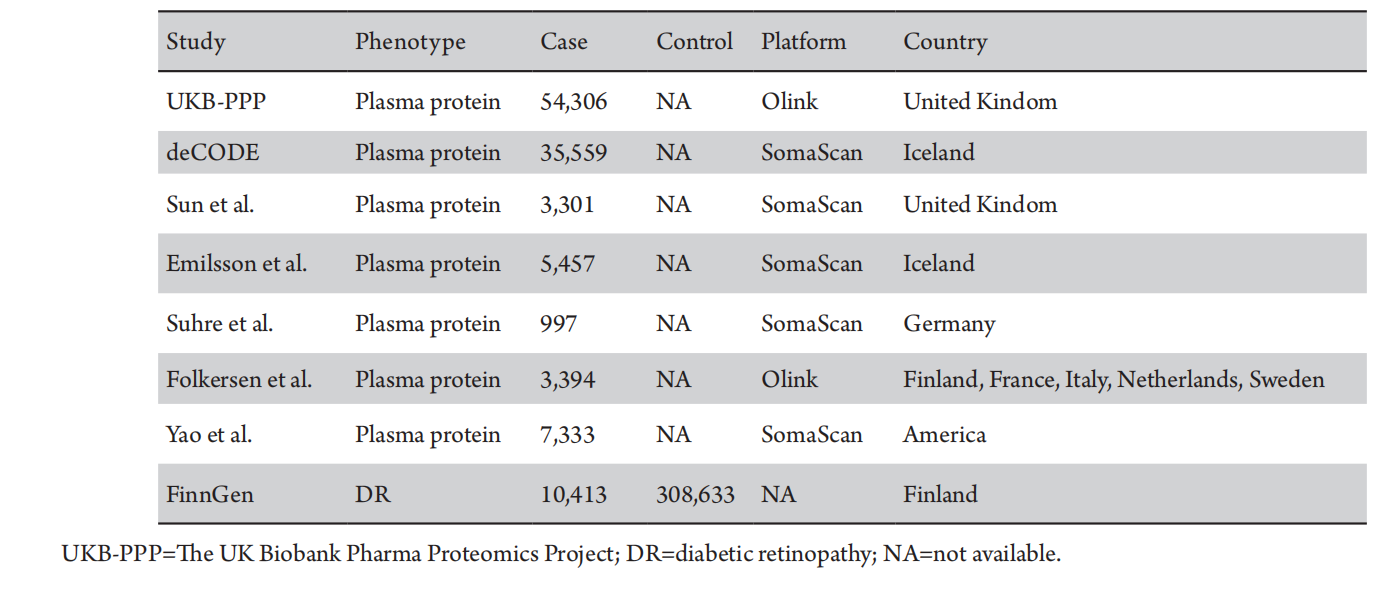
We identified 1,995 cis-pQTLs for 1,954 plasma proteins from the UKB-PPP dataset (Olink platform, n=54,306), and 4,024 cis-pQTLs for 1,380 circulating proteins from the deCODE Health study (Somascan platform, n=35,559). Furthermore, we integrated data on 738 cis-pQTLs linked to 734 plasma proteins, as reported by Zheng et al.[20] These data were derived from GWAS studies utilizing the Somascan platform by Sun et al.,[13] Emilsson et al.,[12] Suhre et al.,[21] and Yao et al.,[11] as well as the GWAS study conducted by Folkersen et al. using the Olink platform (Table 1).
Study Outcome: DR
Data on the plasma protein-related SNPs and DR association were extracted from the FinnGen R9 study, involving 10,413 individuals of European descent as cases and 308,633 control subjects. To ensure the consistency and precision of the clinical endpoint, the diagnostic criteria and levels for DR were constructed from the register codes using the Finnish version of the International Classification of Diseases, 10th revision (ICD-10) diagnosis codes harmonized with definitions from ICD-8 and ICD-9.[22] The whole process has been subjected to multiple evaluations by specialized committees. Stringent protocols were applied to guarantee ethnic homogeneity across the exposure and outcome datasets. Ethical approval and informed consent were acquired from the relevant institutional review boards, acknowledging the study's reliance on established publications and publicly accessible databases.
STATISTICAL ANALYSIS
Two-sample MR Analysis
Genetic tool robustness was assessed through the integration of plasma protein cis-pQTLs from three distinct sources. This involved determining the proportion of variance (R2) explained by the genetic IV for each risk factor and assessing the strength of association (F-statistics) between these genetic tools and the risk factors.[23] To ensure dataset homogeneity, plasma proteins lacking viable genetic tools for the outcome were excluded. An R2 of IV (R2 > 0.1%) identified 6,133 cis-pQTLs representing 2,358 proteins as IV for the analysis.
In the two-sample MR analysis, plasma proteins served as the exposure, with DR as the endpoint. The analysis utilized the "TwoSampleMR" software package. Association estimates between plasma proteins and DR were derived using the Wald ratio (for proteins linked to a single cis-pQTL) and inverse variance-weighted MR (for proteins associated with two or more cis-pQTLs). Confidence intervals (CIs) were computed using the delta method.[24] Results were presented as MR odds ratios (OR) and their corresponding 95% CIs, quantifying the effect of a 1-SD change in gene-predicted plasma protein levels on DR risk. To mitigate the risk of false positives arising from multiple comparisons, the Benjamini and Hochberg correction was applied, setting the False Discovery Rate (FDR) at 0.05. Furthermore, a more stringent correction using the Bonferroni method was implemented, with significance established at P<2.12×10-5 (0.05/2,358). For cis-pQTLs significantly associated with DR in the MR study post-Bonferroni correction, the proportion of variance explained (R2) for these specific proteins was calculated.
Reverse Causality Detection
Bidirectional MR analysis explored the potential reverse causality in the association between DR risk and identified plasma protein levels. Complete GWAS data for proteins associated with DR were sourced from three prior studies. Effect estimates were derived via the MR-IVW method and validated using MR-Egger, weighted median, simple mode, and weighted mode approaches. Statistical significance was set at P<0.05. A Bonferroni correction was applied to account for multiple comparisons, setting significance at P < 2.08 × 10-3.
Bayesian Co-localization Analysis
Co-localization analysis was used to investigate the potential influence of linkage disequilibrium (LD) on the association between identified proteins and DR. This analysis employed a Bayesian model to assign posterior probabilities to five hypotheses related to the presence of a shared variant between the two traits.[25] The hypotheses considered were as follows: (Ⅰ) no causal variant exists for either protein or DR in the genomic locus (H0); (Ⅱ) a causal variant is specific to one protein (H1); (Ⅲ) a causal variant is specific to one DR (H2); (Ⅳ) two distinct causal variants independently affect protein and DR (H3); and (Ⅴ) a shared causal-variant influences both protein and DR (H4). We evaluated the posterior probabilities for H3 (PH3) and H4 (PH4). High-strength co-localization support was indicated when the PH4 was ≥0.8, while moderate-strength co-localization support was defined as 0.5 < PH4 < 0.8. These analyses were conducted using the "coloc" package in R software (version 4.4.1).
MR Assumptions and Phenotype Scanning
MR relies on three foundational assumptions. Firstly, it requires robust associations between SNPs and exposures. Given that all cis-pQTLs exhibited significant associations with their respective protein levels at a genome-wide scale (with P-values < 5 × 10-8 and were identified as the SNPs with the lowest P-values within 1 Mb of the transcriptional start sites of the relevant genes, this assumption is unlikely to pose issues. To gauge the strength of the relationship between genetic instruments and exposure, we computed the F-statistic for the cis-pQTLs, typically exceeding 10 for an SNP to be considered a robust MR tool.
The second MR hypothesis posits that cis-pQTLs should not be linked to a confounding factor in the relationship between exposure and outcome. Ancestry represents a potential source of such confounding, which can be mitigated by ensuring that all participants in the GWAS share European ancestry.
The third MR hypothesis requires that genetic variants do not influence the outcome except through their impact on exposure (horizontal pleiotropy). In this study, potential bias stemming from horizontal pleiotropy is significantly reduced, as the primary MR tool is a cis-acting SNP located within 300 kb of the gene encoding the protein. To explore associations beyond our primary analysis, we employed the 'phenoscanner' software package for a comprehensive scan of previously published large-scale genetic association studies.[26] This scan aimed to unveil potential connections between the identified pQTLs and other traits. SNPs were considered pleiotropic if they met specific criteria: (Ⅰ) their associations achieved genome-wide significance (P < 5 × 10-8); (Ⅱ) the GWAS involved populations of European ancestry; and (Ⅲ) the SNPs exhibited associations with established risk factors for DR, including metabolic traits, proteins, or clinical features. In addition, we calculated the LD r2 between pQTLs for prioritized proteins to unveil potential associations.
Downstream Analysis of Drug Target Proteins
Plasma proteins represent a significant source of potential drug targets. To determine if MR-preferred proteins overlap with genes within the druggable genome, we extracted druggability profiles of target proteins from the updated list of druggable genes.[27] For assessing the clinical development status of drug candidates targeting these proteins, we interrogated the ChEMBL database (version 33) for data on drug molecule classifications, approved indications, and clinical trial outcomes.[28] Furthermore, data regarding drug-target proteins was obtained by searching DrugBank (https://wwws.go.drugbank.com) and manually screening for each candidate target.[29] To assess druggability, proteins were categorized into four groups: approved (for specific proteins with one or more approved drugs); clinical trials (for drugs currently in clinical investigation); preclinical (representing drugs in preclinical development); druggable (relating to proteins designated as potential druggable targets but not found in drug databases).To comprehensively reveal the potential mechanism of MR priority proteins involved in the pathological process of DR, gene function enrichment analysis of all proteins was further completed based on the information from the Gene Ontology database (http://geneontology.org).[30]
Data Availability
Genome-wide summary statistics for Cis-pQTL can be found in the original study,[9, 19-20] while GWAS summary statistics for DR are accessible via the FinnGen consortium website (R9 release) at https://www.finngen.fi/.
RESULTS
Figure 1 presents the framework of this study. The three separate datasets consolidated cis-pQTL data pertaining to plasma proteins, resulting in the discovery of 6,362 cis-pQTLs associated with 2,602 unique proteins (Table 1). The subsequent MR analysis incorporated 2,358 proteins from the FinnGen R9 dataset, following a rigorous screening process for genetic instrument tools with an R2 exceeding 0.01%, and the exclusion of plasma proteins lacking suitable genetic instruments for further analysis.
Table 1 Data sources for studied phenotypes
Genetic Association Between Plasma Proteins and DR
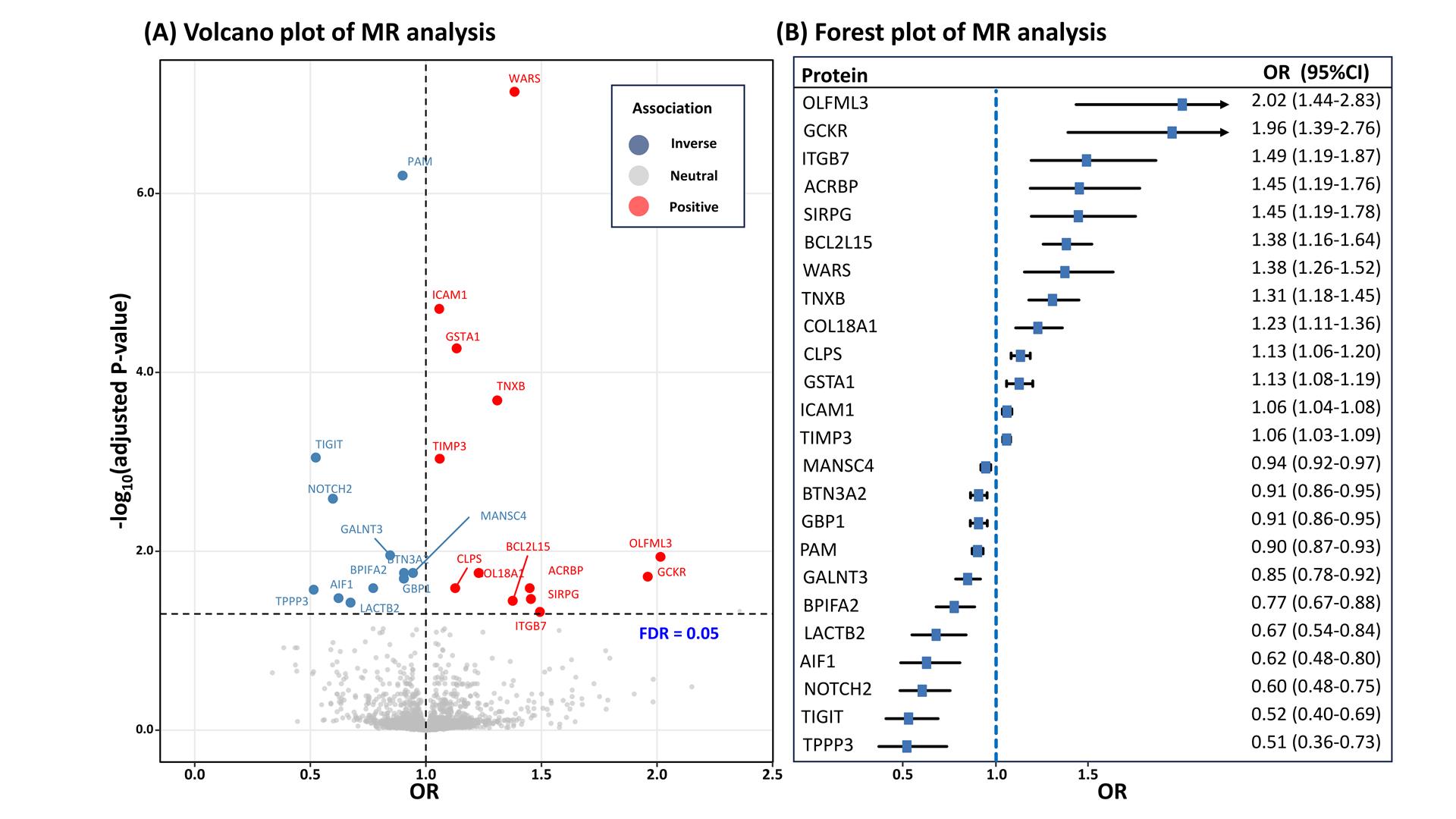
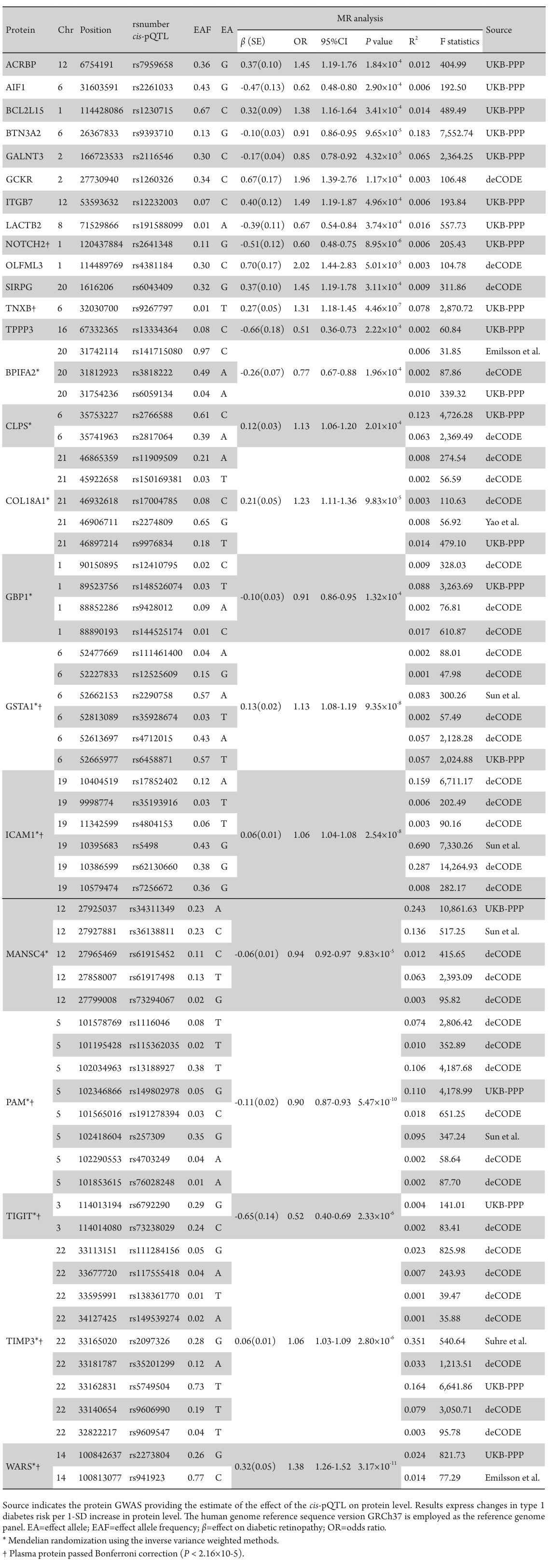
Using 6,362 cis-pQTLs as genetic instruments for the corresponding proteins, the MR analysis unveiled associations between 24 plasma proteins and DR risk, following FDR correction (Table 2). With each SD increase in genetically predicted protein levels, the OR for DR spanned from 0.51(95% CI: 0.36-0.73; P=2.22×10-4) for tubulin polymerization-promoting protein family member 3 (TPPP3), to 2.02 (95% CI: 1.44-2.83; P=5.01×10-5) for olfactomedin like 3 (OLFML3). There are 11 proteins positively associated with DR risk and 13 proteins inversely associated (Table 2, Figure 2). After Bonferroni correction (P < 2.16×10-5), the MR analysis sustained significant associations for 8 proteins with DR risk, with all causal proteins displaying F-statistic values exceeding 10, excluding bias from weak instrumental variables.
Figure 2 Result summary of MR and colocalization analysis on the associations between plasma proteins and the risk of diabetic retinopathy. OR=odds ratio; FDR=false discovery rate
Table 2 Plasma proteins significantly associated with diabetic retinopathy after false discovery rate (FDR) correction
Sensitivity Analyses
To evaluate the robustness of estimates derived from primary MR analyses, multiple sensitivity analyses were conducted on the 24 causal proteins. In the bidirectional MR analysis, after applying a stringent Bonferroni correction (P < 2.08 × 10-3), none of the causal plasma proteins showed signs of reverse causation (Table 3).
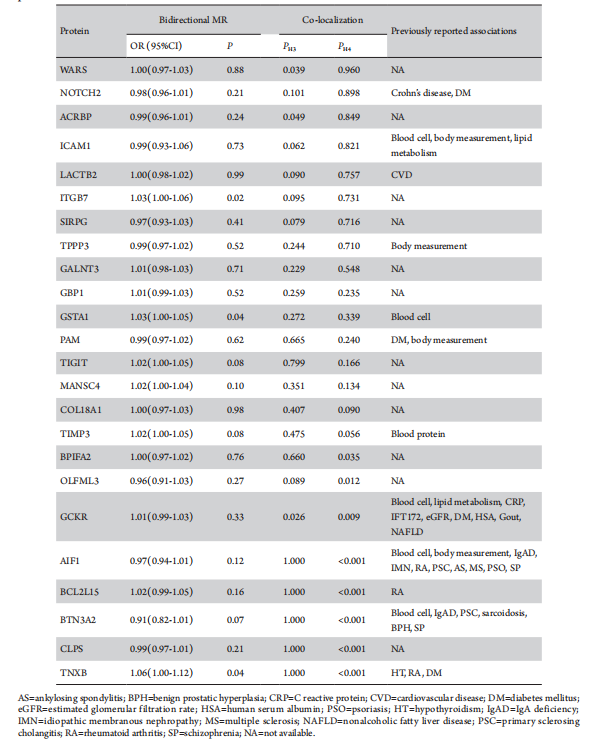
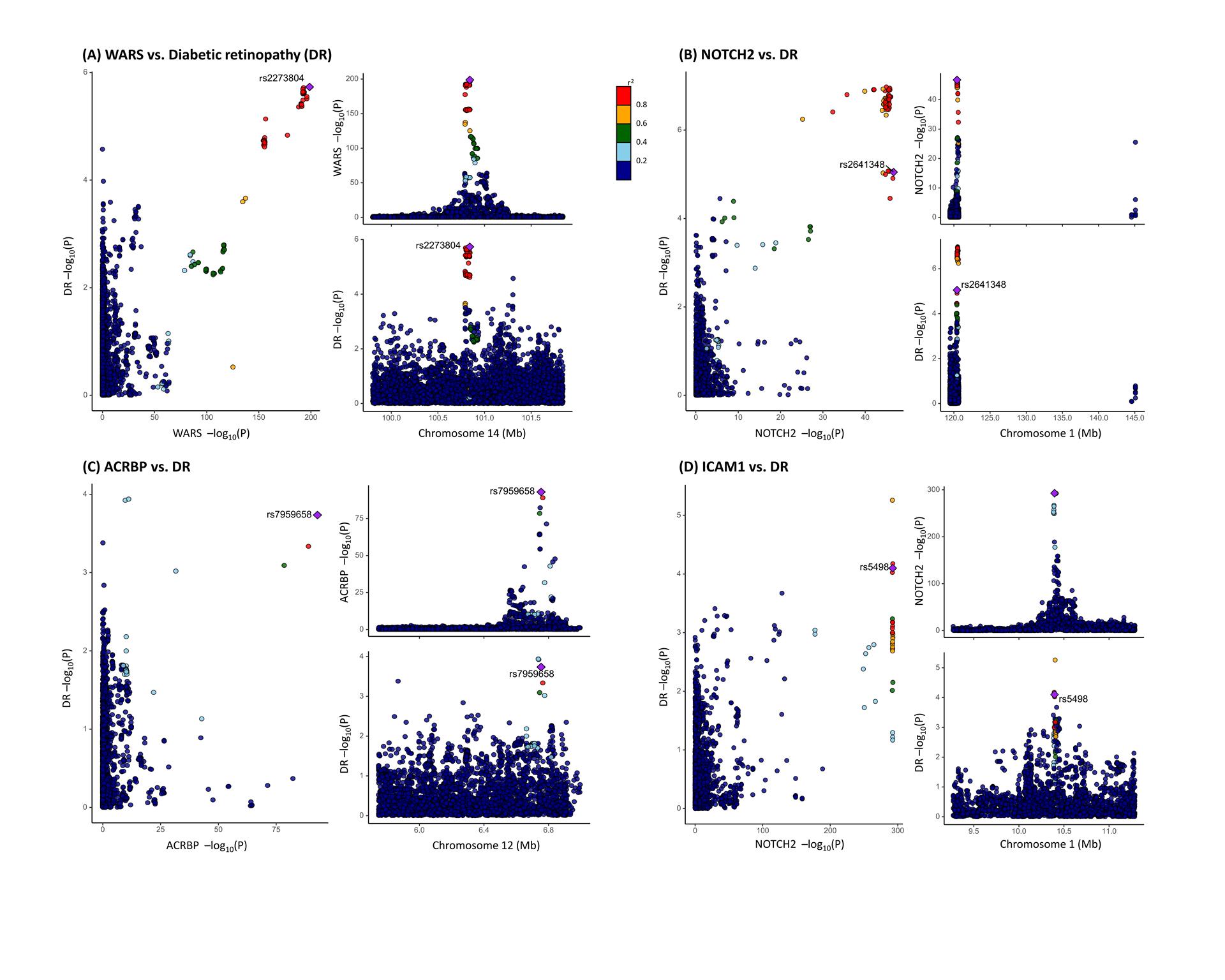
To examine potential confounding due to LD, colocalization analysis was conducted to assess whether genetic factors influencing plasma protein levels intersected causally with those affecting DR. Among the 24 DR-associated proteins, acrosin-binding protein (ACRBP), intercellular adhesion molecule 1 (ICAM1), neurogenic locus notch homolog protein 2 (NOTCH2), and cytoplasmic tRNA synthetase (WARS) exhibited strong evidence of co-localization (PH4≥0.8). The endoribonuclease LACTB2 (LACTB2), integrin beta-7 (ITGB7), signal regulatory protein gamma (SIRPG), tubulin polymerization-promoting protein family member 3 (TPPP3), and polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3) demonstrated moderate-strength evidence of co-localization analysis (0.8 > PH4≥0.5) (Table 3; Figure 3). These findings suggest a potential shared pool of genetic variants between these proteins and DR.
The phenotypic scan of cis-pQTLs linked to all DR-associated proteins revealed connections of CGKR (rs1260326), NOTCH2 (rs2641348), PAM (rs1116046), and TNXB (rs9267797) with diabetes mellitus. However, no such associations were detected between these cis-pQTLs and DR (Table 3). In addition, the phenotypic scan unveiled correlations of specific DR-associated proteins, including AIF1 (rs2261033), BTN3A2 (rs9393710), GCKR (rs1260326), GSTA1 (rs2290758), ICAM1 (rs5498), and TIMP3 (2097326), with various blood constituents. Particular plasma protein cis-pQTLs displayed links to various diseases, such as IgA deficiency, idiopathic membranous nephropathy, rheumatoid arthritis, primary sclerosing cholangitis, ankylosing spondylitis, multiple sclerosis, psoriasis, schizophrenia, sarcoidosis, benign prostatic hyperplasia, and schizophrenia, gout, nonalcoholic fatty liver disease. LACTB2 (rs191588099) showed a connection with cerebrovascular disease, NOTCH2 (rs2641348) with Crohn's disease, and TNXB (rs9267797) with hypothyroidism and rheumatoid arthritis (Table 3). Relaxing the association significance threshold to (P<1×10-5) revealed no cis-pQTLs for any plasma proteins maintaining associations with DR, thus affirming the robustness of the MR analysis results in the presence of potential confounding factors.
Table 3 Summary of reverse causality detection, Bayesian co-localization analysis and phenotype scanning on 24 potential causal proteins
Figure 3 Co-localization analysis of the association between plasma protein level and diabetic retinopathy (DR) (A) Co-localization analysis of the association between WARS level and DR: the cis-pQTL rs2273804 is shown as a purple diamond. The P values of the SNPs in the WARS locus were extracted from the GWAS by UKB-PPP and from the diabetic retinopathy GWAS by the FinnGen study. (B) Co-localization analysis of the association between NOTCH2 level and DR: the cis-pQTL rs2641348 is shown as a purple diamond. The P values of the SNPs in the NOTCH2 locus were extracted from the GWAS by UKB-PPP and from the GWAS by FinnGen study. (C) Co-localization analysis of the association between ACRBP level and DR: the cis-pQTL rs7959658 is shown as a purple diamond. The P values of the SNPs in the ACRBP locus were extracted from the GWAS by UKB-PPP and from the GWAS by FinnGen study. (D) Co-localization analysis of the association between ICAM1 level and DR: the cis-pQTL rs5498 is shown as a purple diamond. The P values of the SNPs in the ICAM1 locus were extracted from the GWAS by deCODE HEALTH and from the GWAS by FinnGen study. The dots in the scatter plots are colored according to their LD to the identified casual cis-pQTL. LD is calculated based on the 1000 Genomes phase 3 European references. Mb=Megabase pairs.
Downstream Analysis of MR-derived Protein Targets
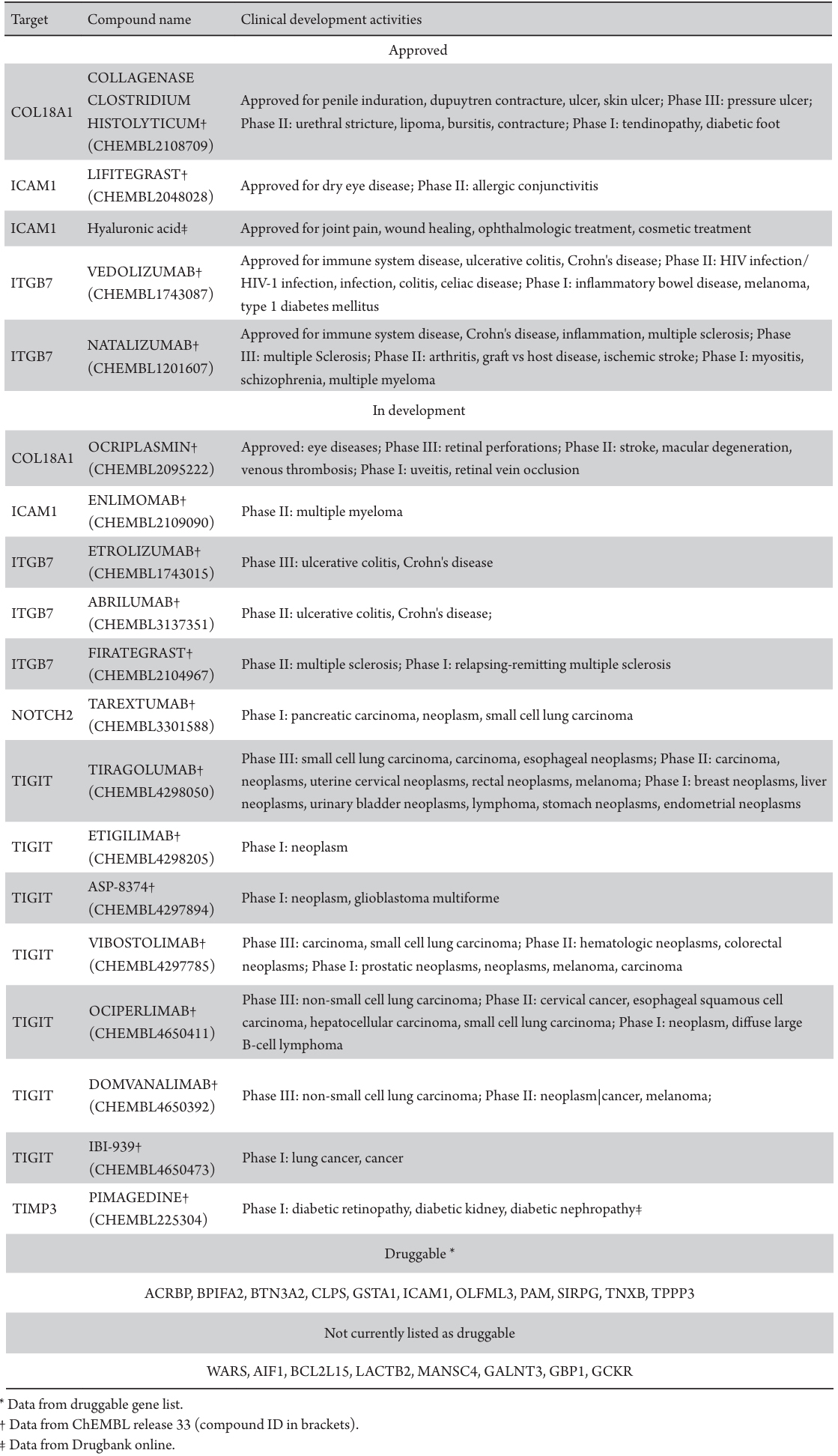
To evaluate the druggability and developmental status of plasma protein targets associated with DR, an extensive investigation utilizing druggable gene lists, ChEMBL (release 33) drug discovery database, and DrugBank (release 5.1.10) was conducted. These targets were categorized into three groups based on their progress in drug development: approved (targets of drugs already approved for one or more conditions), in development (currently in clinical trials), and druggable (potential targets;Table 4). The analysis of drug development activities identified six plasma proteins (COL18A1, ICAM1, ITGB7, NOTCH2, TIGHT, and TIMP3) as the focus of drug development endeavors.
Table 4 Summary of druggability and clinical development activity for diabetic retinopathy associated with causal associations on MR analysis
COL18A1 was targeted by drugs approved for various conditions, including penile induration, dupuytren contracture, ulcer, skin ulcer (e.g., collagenase clostridium histolyticum), and eye diseases (e.g., ocriplasmin). Similarly, ICAM1 was a subject of drug development for dry eye disease (e.g., lifitegrast) and other conditions like joint pain, wound healing, ophthalmologic and cosmetic treatments (e.g., hyaluronic acid). ITGB7 attracted attention from drugs such as vedolizumab and natalizumab, addressing immune system diseases, ulcerative colitis, Crohn's disease, inflammation, and multiple sclerosis. Among the six DR-related plasma proteins, three have associations with six approved drugs, and all six of them are linked to one or more drugs undergoing Phase I-III clinical trials. Notably, pimagedine, targeting TIMP3, was in Phase I trials for diabetic complications, including DR. However, there was no evidence of drug development activities for the remaining 18 plasma proteins. Based on the list of druggable genes, ACRBP, BPIFA2, BTN3A2, CLPS, GSTA1, OLFML3, PAM, SIRPG, and TNXB were identified as druggable genes, offering potential targets for future drug development initiatives.
GO enrichment analysis revealed functional pathways enriched by genes fornovel drug target proteins. TheSupplementary Figure shows that 18 out of 24 plasma proteins are involved in 22 gene functional pathways. These pathways include epithelial cell differentiation, positive regulation of cytokine production, supramolecular fiber organization, negative regulation of cell population proliferation, and blood vessel development, among others.
DISCUSSION
Summary of the Findings
This first PW-MR study successfully identified 24 DR causal plasma proteins with potential for drug targeting, comprising 19 novel DR susceptibility proteins and five previously known (COL18A1, GBP1, ICAM1, NOTCH2, TIMP3) proteins. Co-localization analysis revealed that genetically higher levels of WARS, ACRBP, and ICAM1 were inversely related to DR risk, while elevated levels of NOTCH2 increased DR risk. Three of these 24 proteins are being developed as therapeutic agents for dry eye disease and an immune disorder, with two additional proteins undergoing phase I clinical trials as targeted treatments for DR. This study reinforced the strategies for DR drug development, and underscored the potential of PW-MR for unraveling the causal biology of complex diseases.
MR-derived Novel Biomarkers and Potential Targets for DR
This study identified 19 novel drug target proteins for DR, notably focusing on OLFML3, ACRBP,WARS, and TPPP3. Among them, ACRBP and WARS demonstrate significant potential in their involvement with the pathogenesis of DR, supported by strong evidence of colocalization. Acrosin-binding protein (ACRBP) is recognized as a glycoprotein that plays a pivotal role in regulating protease activity during sperm penetration through the zona pellucida.[31] Recent studies have shown that ACRBP is expressed abnormally in various types of tumors,[32] which suggests that ACRBP may counteract pathological interferences with microtubule dynamics and centrosome functions, ensuring normal mitosis during angiogenesis.[33] This study suggests that ACRBP may be a causative protein in the development of DR at the genetic level, suggesting the need to further explore specific drug-targeting mechanisms. Additionally, tryptophanyl-tRNA synthetase (WARS) is an essential housekeeping enzyme that attaches tryptophan to its corresponding tRNA for protein synthesis.[34] Studies have shown a positive correlation between WARS and the expression of T-cell markers,[35] and the anterior segment of the retina in patients with DR has been found to contain numerous immune-inflammatory cells, including T-cells.[36] Based onprevious findings, this study further supports the notion that WARS is a plasma protein that increases the risk of DR. However, these preliminary insights necessitate further validation through comprehensive experimental and clinical studies.
Compared to Previous Experimental and Observational Studies
Among the proteins identified as causal through MR, 6 have been indicated by previous studies. Intercellular adhesion molecule 1 (ICAM1), critical for leukocyte adhesion and migration, consistently exhibits higher levels in DR patients than in controls, a finding substantiated by a meta-analysis of 11 studies, indicating a potential correlation with DR severity.[37] Vascular endothelial growth factor, a key element in retinopathy pathogenesis, promotes the release of serum sICAM1 from ICAM1's outer segment, intensifying inflammation and DR progression.[37-38] This study underscored the causal relationship between increased ICAM1 levels and DR risk, with drugs targeting ICAM1, such as LIFITEGRAST, having gained FDA approval for clinical treatment of dry eye disease. NOTCH signaling, is crucial for retinal vascular development and eye diseases, but previous studies show conflicting outcomes, particularly regarding NOTCH2.[39-40] This study revealed that genetic NOTCH2 levels confer protection against DR, underscoring the need for further studies to elucidate the precise nature of this association.
Aligns with MR Assumptions and Co-localization Evidence
To enhance the robustness and reliability of the findings, a comprehensive method was implemented, ensuring adherence to the MR assumptions. Firstly, we focused on selecting cis-pQTLs due to their inherently higher biological likelihood of influencing protein expression in nearby genes, thus significantly affecting protein function and activity, compared to trans-pQTLs. The selection criteria for instrumental variables for plasma protein levels included LD clustering with an r2 below 0.001, cis-pQTLs with a P-value of 5.0 × 10-8 or less, and a location within 1 Mb of the relevant protein-coding gene. We also assessed the strength of these genetic IVs, requiring F-statistics greater than 10, to reduce bias from weaker instrumental variables. Secondly, utilizing an integrated analysis that combined MR and co-localization, we identified novel drug targets for DR. Thirdly, we conducted a bidirectional MR analysis to address potential reverse causality and genetic confounding linked to LD. Furthermore, Bayesian co-localization analysis effectively ruled out confounding by LD. This led to the identification of four proteins (WARS, NOTCH2, ACRBP, ICAM1) as likely sharing genetic variants with DR. However, non-co-localization does not undermine these findings, given the high false-negative rate (approximately 60%) associated with Bayesian co-localization methods. Phenotypic scanning also linked two proteins (ACRBP, LACTB2) with other traits, such as blood cells and body measurements, but these links do not fully clarify their relationship with DR. Therefore, WARS, NOTCH2, ACRBP, and ICAM1 are promising drug targets for DR.
Scientific and Clinical Implications
This study had crucial insights for scientific research and clinical practice. Firstly, it underscores the potential of expanding omic biomarker testing in discovering novel drug targets. By aggregating genetic data from extensive plasma proteomics studies, we analyzed 2,602 circulating proteins related to DR, representing a fraction (10%) of all known proteins. Secondly, our PW-MR findings suggest that proteins causally associated with DR may also modulate the risk of various non-DR diseases. Several proteins identified are either approved for other conditions or in clinical trials, suggesting a potential overlap in causal proteins across different diseases. Thirdly, PW-MR stands out as an effective preclinical approach for prioritizing drug targets and anticipating target-related side effects. The ongoing exploration of these biomarkers is crucial, promising to unveil immediate mediators and enhance the effectiveness and safety of emerging drug targets.
Strength and limitations
The strength of this study lies in using the most recent, comprehensive plasma protein dataset and the largest GWAS dataset for DR, thereby improving our ability to detect associations. By focusing on individuals of European ancestry, we minimized population stratification bias. Our findings are validated through various sensitivity analyses, including bidirectional MR and co-localization analyses, which are crucial for identifying shared genetic variants between exposure outcomes. This study also has several limitations. Firstly, genetic variants and qualitative protein changes, such as amino acid substitutions and post-translational modifications, may compromise the specificity of protein quantification. These changes can affect binding affinity in assays, potentially leading to misinterpretations.[41] To address this, we adopted multiple protein quantification methods including Olink, Somascan, and various genetic IVs, thereby minimizing the impact of technology-specific biases on our results. Secondly, the exclusive focus on cis-acting SNPs limited our choice of analytical methods. However, our meticulous selection of SNPs from multiple studies allowed for the use of robust IVs. The study's scope was confined to proteins associated with identifiable cis-pQTL signaling, possibly overlooking other relevant therapeutic targets. Despite this, we conducted a comprehensive co-localization of circulating proteins to identify potential causal genes for DR. Thirdly, our standardized units of protein expression do not directly convey the absolute impact on DR, nor facilitate direct comparisons across proteins.[42] However, the inferred causal directions provide valuable insights for predicting efficacy and side effects, which can be substantiated in future clinical trials. Finally, the inclusion of only the European population limits the applicability of our findings to other ethnic groups. This highlights the need for additional studies to validate these results in more diverse populations.
CONCLUSION
In summary, we leveraged two-sample MR analysis using extensive proteomic data to identify 24 potential drug targets for diabetic retinopathy (DR). Four plasma proteins (WARS, ACRBP, ICAM1, and NOTCH2) emerged as particularly promising candidates, supported by robust co-localization evidence. This study offers unprecedented insights into DR’s etiology and potential therapeutic strategies. It also highlights how burgeoning genomic and proteomic datasets can be pivotal in identifying novel drug targets. Further experimental and clinical studies are warranted to evaluate the utility and efficacy of these candidate proteins.
Acknowledgments
This study was funded by the Hainan Province Clinical Medical Center(82171084), and the National Natural Science Foundation of China (82371086).
Author contributions statement
Study concept and design: W.W., W.H.; Acquisition, analyses, or interpretation: All authors; Drafting of the manuscript: S.Y., W.W., Z.Z.; Critical revision of the manuscript for important intellectual content: All authors; Statistical analyses: W.W., S.Y., Z.Z.; Obtained funding: W.W., W.H.; Administrative, technical, or material support: S.Y., Z.Z., W.H.; Study supervision: W.W., W.H.
Ethics approval
The study is based on publicly available data from large-scale genome-wide association studies. Included studies had been approved by corresponding ethical review committees.
Competing interest statement
No potential conflicts of interest relevant to this article were reported.
Data sharing statement
All data analyzed in this study can be obtained by a reasonable request to the corresponding authors.
1、Teo ZL, Tham YC, Yu M, et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021; 128(11): 1580-91.Teo ZL, Tham YC, Yu M, et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021; 128(11): 1580-91.
2、Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010; 376(9735): 124-36.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010; 376(9735): 124-36.
3、Boyer DS, Yoon YH, Belfort RJ, et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014; 121(10): 1904-14.Boyer DS, Yoon YH, Belfort RJ, et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014; 121(10): 1904-14.
4、Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1(11): 845-67.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1(11): 845-67.
5、Chouliaras L, Thomas A, Malpetti M, et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer's disease, frontotemporal dementia and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2022; 93(6): 651-8.Chouliaras L, Thomas A, Malpetti M, et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer's disease, frontotemporal dementia and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2022; 93(6): 651-8.
6、Chan MY, Efthymios M, Tan SH, et al. Prioritizing Candidates of Post-Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020; 142(15): 1408-21.Chan MY, Efthymios M, Tan SH, et al. Prioritizing Candidates of Post-Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020; 142(15): 1408-21.
7、Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov 2017; 16(1): 19-34.Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov 2017; 16(1): 19-34.
8、Pietzner M, Wheeler E, Carrasco-Zanini J, et al. Mapping the proteo-genomic convergence of human diseases. Science 2021; 374(6569): eabj1541.Pietzner M, Wheeler E, Carrasco-Zanini J, et al. Mapping the proteo-genomic convergence of human diseases. Science 2021; 374(6569): eabj1541.
9、Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet 2021; 53(12): 1712-21.Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet 2021; 53(12): 1712-21.
10、Ursu O, Glick M, Oprea T. Novel drug targets in 2018. Nat Rev Drug Discov 2019.Ursu O, Glick M, Oprea T. Novel drug targets in 2018. Nat Rev Drug Discov 2019.
11、Yao C, Chen G, Song C, et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun 2018; 9(1): 3268.Yao C, Chen G, Song C, et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun 2018; 9(1): 3268.
12、Emilsson V, Ilkov M, Lamb JR, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018; 361(6404): 769-73.Emilsson V, Ilkov M, Lamb JR, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018; 361(6404): 769-73.
13、Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature 2018; 558(7708): 73-9.Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature 2018; 558(7708): 73-9.
14、Hulur I, Gamazon ER, Skol AD, et al. Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genomics 2015; 16(1): 138.Hulur I, Gamazon ER, Skol AD, et al. Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genomics 2015; 16(1): 138.
16、Malarstig A, Grassmann F, Dahl L, et al. Evaluation of circulating plasma proteins in breast cancer using Mendelian randomisation. Nat Commun 2023; 14(1): 7680.Malarstig A, Grassmann F, Dahl L, et al. Evaluation of circulating plasma proteins in breast cancer using Mendelian randomisation. Nat Commun 2023; 14(1): 7680.
17、Chen L, Peters JE, Prins B, et al. Systematic Mendelian randomization using the human plasma proteome to discover potential therapeutic targets for stroke. Nat Commun 2022; 13(1): 6143.Chen L, Peters JE, Prins B, et al. Systematic Mendelian randomization using the human plasma proteome to discover potential therapeutic targets for stroke. Nat Commun 2022; 13(1): 6143.
18、Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015; 47(8): 856-60.Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015; 47(8): 856-60.
19、Sun BB, Chiou J, Traylor M, et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 2023; 622(7982): 329-38.Sun BB, Chiou J, Traylor M, et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 2023; 622(7982): 329-38.
20、Zheng J, Haberland V, Baird D, et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat Genet 2020; 52(10): 1122-31.Zheng J, Haberland V, Baird D, et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat Genet 2020; 52(10): 1122-31.
21、Suhre K, Arnold M, Bhagwat AM, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun 2017; 8: 14357.Suhre K, Arnold M, Bhagwat AM, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun 2017; 8: 14357.
22、Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023; 613(7944): 508-18.Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023; 613(7944): 508-18.
23、Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011; 40(3): 755-64.Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011; 40(3): 755-64.
24、Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 2013; 178(7): 1177-84.Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 2013; 178(7): 1177-84.
25、Foley CN, Staley JR, Breen PG, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun 2021; 12(1): 764.Foley CN, Staley JR, Breen PG, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun 2021; 12(1): 764.
26、Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019; 35(22): 4851-3.Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019; 35(22): 4851-3.
27、Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017; 9(383).Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017; 9(383).
28、Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019; 47(D1): D930-40.Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019; 47(D1): D930-40.
29、Knox C, Wilson M, Klinger CM, et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res 2023.Knox C, Wilson M, Klinger CM, et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res 2023.
30、Mi H, Muruganujan A, Huang X, et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 2019; 14(3): 703-21.Mi H, Muruganujan A, Huang X, et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 2019; 14(3): 703-21.
31、Mohaqiq M, Movahedin M, Mazaheri Z, Amirjannati N. In vitro transplantation of spermatogonial stem cells isolated from human frozen-thawed testis tissue can induce spermatogenesis under 3-dimensional tissue culture conditions. Biol Res 2019; 52(1): 16.Mohaqiq M, Movahedin M, Mazaheri Z, Amirjannati N. In vitro transplantation of spermatogonial stem cells isolated from human frozen-thawed testis tissue can induce spermatogenesis under 3-dimensional tissue culture conditions. Biol Res 2019; 52(1): 16.
32、Tammela J, Uenaka A, Ono T, et al. OY-TES-1 expression and serum immunoreactivity in epithelial ovarian cancer. Int J Oncol 2006; 29(4): 903-10.Tammela J, Uenaka A, Ono T, et al. OY-TES-1 expression and serum immunoreactivity in epithelial ovarian cancer. Int J Oncol 2006; 29(4): 903-10.
33、Whitehurst AW, Xie Y, Purinton SC, et al. Tumor antigen acrosin binding protein normalizes mitotic spindle function to promote cancer cell proliferation. Cancer Res 2010; 70(19): 7652-61.Whitehurst AW, Xie Y, Purinton SC, et al. Tumor antigen acrosin binding protein normalizes mitotic spindle function to promote cancer cell proliferation. Cancer Res 2010; 70(19): 7652-61.
34、Jin M. Unique roles of tryptophanyl-tRNA synthetase in immune control and its therapeutic implications. Exp Mol Med 2019; 51(1): 1-10.Jin M. Unique roles of tryptophanyl-tRNA synthetase in immune control and its therapeutic implications. Exp Mol Med 2019; 51(1): 1-10.
35、Ahn YH, Oh SC, Zhou S, Kim TD. Tryptophanyl-tRNA Synthetase as a Potential Therapeutic Target. Int J Mol Sci 2021; 22(9).Ahn YH, Oh SC, Zhou S, Kim TD. Tryptophanyl-tRNA Synthetase as a Potential Therapeutic Target. Int J Mol Sci 2021; 22(9).
37、Yao Y, Du J, Li R, et al. Association between ICAM-1 level and diabetic retinopathy: a review and meta-analysis. Postgrad Med J 2019; 95(1121): 162-8.Yao Y, Du J, Li R, et al. Association between ICAM-1 level and diabetic retinopathy: a review and meta-analysis. Postgrad Med J 2019; 95(1121): 162-8.
38、Funatsu H, Yamashita H, Sakata K, et al. Vitreous levels of vascular endothelial growth factor and intercellular adhesion molecule 1 are related to diabetic macular edema. Ophthalmology 2005; 112(5): 806-16.Funatsu H, Yamashita H, Sakata K, et al. Vitreous levels of vascular endothelial growth factor and intercellular adhesion molecule 1 are related to diabetic macular edema. Ophthalmology 2005; 112(5): 806-16.
39、Wang N, Ding L, Liu D, et al. Molecular investigation of candidate genes for pyroptosis-induced inflammation in diabetic retinopathy. Front Endocrinol (Lausanne) 2022; 13: 918605.Wang N, Ding L, Liu D, et al. Molecular investigation of candidate genes for pyroptosis-induced inflammation in diabetic retinopathy. Front Endocrinol (Lausanne) 2022; 13: 918605.
40、Dou GR, Wang L, Wang YS, Han H. Notch signaling in ocular vasculature development and diseases. Mol Med 2012; 18(1): 47-55.Dou GR, Wang L, Wang YS, Han H. Notch signaling in ocular vasculature development and diseases. Mol Med 2012; 18(1): 47-55.
41、Zhang J, Zeng Y, Chen J, et al. miR-29a/b cluster suppresses high glucose-induced endothelial-mesenchymal transition in human retinal microvascular endothelial cells by targeting Notch2. Exp Ther Med 2019; 17(4): 3108-16.Zhang J, Zeng Y, Chen J, et al. miR-29a/b cluster suppresses high glucose-induced endothelial-mesenchymal transition in human retinal microvascular endothelial cells by targeting Notch2. Exp Ther Med 2019; 17(4): 3108-16.
42、Joshi A, Mayr M. In Aptamers They Trust: The Caveats of the SOMAscan Biomarker Discovery Platform from SomaLogic. Circulation 2018; 138(22): 2482-5.Joshi A, Mayr M. In Aptamers They Trust: The Caveats of the SOMAscan Biomarker Discovery Platform from SomaLogic. Circulation 2018; 138(22): 2482-5.
43、Henry A, Gordillo-Maranon M, Finan C, et al. Therapeutic Targets for Heart Failure Identified Using Proteomics and Mendelian Randomization. Circulation 2022; 145(16): 1205-17.Henry A, Gordillo-Maranon M, Finan C, et al. Therapeutic Targets for Heart Failure Identified Using Proteomics and Mendelian Randomization. Circulation 2022; 145(16): 1205-17.