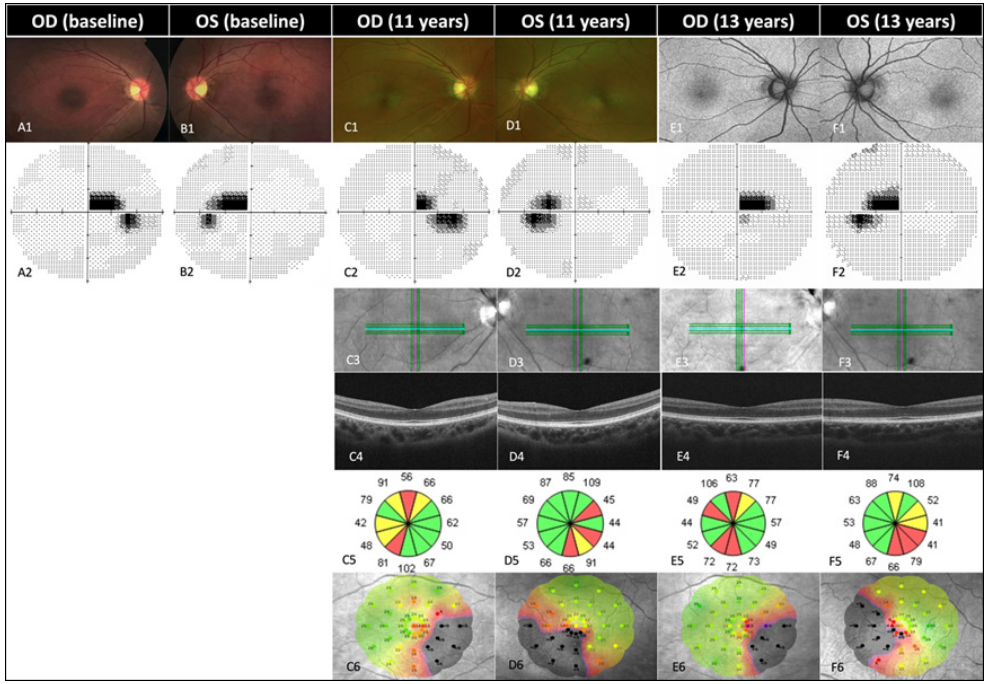
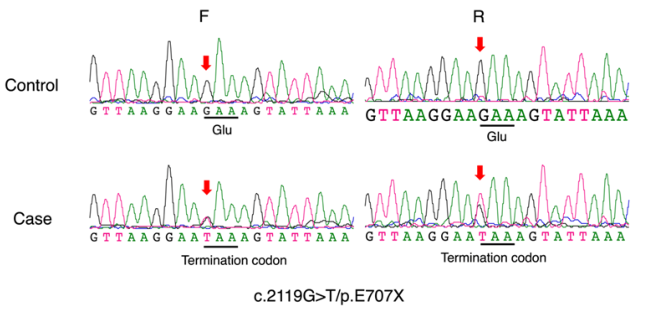
1、Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. 1959, 164(Supp 54): 1-147.Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. 1959, 164(Supp 54): 1-147.
2、Lenaers G, Hamel C, Delettre C, et al. Dominant optic atrophy. Orphanet J Rare Dis. 2012, 7: 46. DOI: 10.1186/1750-1172-7-46.Lenaers G, Hamel C, Delettre C, et al. Dominant optic atrophy. Orphanet J Rare Dis. 2012, 7: 46. DOI: 10.1186/1750-1172-7-46.
3、Chen Y, Jia X, Wang P, et al. Mutation survey of the optic atrophy 1 gene in 193 Chinese families with suspected hereditary optic neuropathy. Mol Vis. 2013, 19: 292-302.Chen Y, Jia X, Wang P, et al. Mutation survey of the optic atrophy 1 gene in 193 Chinese families with suspected hereditary optic neuropathy. Mol Vis. 2013, 19: 292-302.
4、Skidd PM, Lessell S, Cestari DM. Autosomal dominant hereditary optic neuropathy (ADOA): a review of the genetics and clinical manifestations of ADOA and ADOA+. Semin Ophthalmol. 2013, 28(5-6): 422-426. DOI: 10.3109/08820538.2013.825296.Skidd PM, Lessell S, Cestari DM. Autosomal dominant hereditary optic neuropathy (ADOA): a review of the genetics and clinical manifestations of ADOA and ADOA+. Semin Ophthalmol. 2013, 28(5-6): 422-426. DOI: 10.3109/08820538.2013.825296.
5、Yu-Wai-Man P, Griffiths PG, Burke A, et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010, 117(8): 1538-1546, 1546.e1. DOI: 10.1016/j.ophtha.2009.12.038.Yu-Wai-Man P, Griffiths PG, Burke A, et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010, 117(8): 1538-1546, 1546.e1. DOI: 10.1016/j.ophtha.2009.12.038.
6、Aleo SJ, Del Dotto V, Fogazza M, et al. Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum Mol Genet. 2021, 29(22): 3631-3645. DOI: 10.1093/hmg/ddaa244.Aleo SJ, Del Dotto V, Fogazza M, et al. Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum Mol Genet. 2021, 29(22): 3631-3645. DOI: 10.1093/hmg/ddaa244.
7、Payne M, Yang Z, Katz BJ, et al. Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: a syndrome caused by a missense mutation in OPA1. Am J Ophthalmol. 2004, 138(5): 749-755. DOI: 10.1016/ j.ajo.2004.06.011.Payne M, Yang Z, Katz BJ, et al. Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: a syndrome caused by a missense mutation in OPA1. Am J Ophthalmol. 2004, 138(5): 749-755. DOI: 10.1016/ j.ajo.2004.06.011.
8、Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi- system neurological disease is common in patients with OPA1 mutations. Brain. 2010, 133(Pt 3): 771-786. DOI: 10.1093/brain/awq007.Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi- system neurological disease is common in patients with OPA1 mutations. Brain. 2010, 133(Pt 3): 771-786. DOI: 10.1093/brain/awq007.
9、Reynier P, Amati-Bonneau P, Verny C, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. 2004, 41(9): e110. DOI: 10.1136/jmg.2003.016576.Reynier P, Amati-Bonneau P, Verny C, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. 2004, 41(9): e110. DOI: 10.1136/jmg.2003.016576.
10、Del%20Dotto%20V%2C%20Fogazza%20M%2C%20Lenaers%20G%2C%20et%20al.%20OPA1%3A%20how%20much%20do%20we%20know%20to%20approach%20therapy%3F%20Pharmacol%20Res.%202018%2C%20131%3A%20199-210.%20DOI%3A%2010.1016%2Fj.phrs.2018.02.018.Del%20Dotto%20V%2C%20Fogazza%20M%2C%20Lenaers%20G%2C%20et%20al.%20OPA1%3A%20how%20much%20do%20we%20know%20to%20approach%20therapy%3F%20Pharmacol%20Res.%202018%2C%20131%3A%20199-210.%20DOI%3A%2010.1016%2Fj.phrs.2018.02.018.
11、von der Malsburg A, Sapp GM, Zuccaro KE, et al. Structural mechanism of mitochondrial membrane remodelling by human OPA1. Nature. 2023, 620(7976): 1101-1108. DOI: 10.1038/s41586-023-06441-6.von der Malsburg A, Sapp GM, Zuccaro KE, et al. Structural mechanism of mitochondrial membrane remodelling by human OPA1. Nature. 2023, 620(7976): 1101-1108. DOI: 10.1038/s41586-023-06441-6.
12、Nyenhuis SB, Wu X, Strub MP, et al. OPA1 helical structures give perspective to mitochondrial dysfunction. Nature. 2023, 620(7976): 1109-1116. DOI: 10.1038/ s41586-023-06462-1.Nyenhuis SB, Wu X, Strub MP, et al. OPA1 helical structures give perspective to mitochondrial dysfunction. Nature. 2023, 620(7976): 1109-1116. DOI: 10.1038/ s41586-023-06462-1.
13、Olichon A, Landes T, Arnauné-Pelloquin L, et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. 2007, 211(2): 423-430. DOI: 10.1002/jcp.20950.Olichon A, Landes T, Arnauné-Pelloquin L, et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. 2007, 211(2): 423-430. DOI: 10.1002/jcp.20950.
14、Weisschuh N, Schimpf-Linzenbold S, Mazzola P, et al. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants. PLoS One. 2021, 16(7): e0253987. DOI: 10.1371/journal. pone.0253987.Weisschuh N, Schimpf-Linzenbold S, Mazzola P, et al. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants. PLoS One. 2021, 16(7): e0253987. DOI: 10.1371/journal. pone.0253987.
15、Rosenthal AR, Kolb H, Bergsma D, et al. Chloroquine retinopathy in the rhesus monkey. Invest Ophthalmol Vis Sci. 1978, 17(12): 1158-1175.Rosenthal AR, Kolb H, Bergsma D, et al. Chloroquine retinopathy in the rhesus monkey. Invest Ophthalmol Vis Sci. 1978, 17(12): 1158-1175.
16、Latasiewicz M, Gourier H, Yusuf IH, et al. Hydroxychloroquine retinopathy: an emerging problem. Eye, 2017, 31(6): 972-976. DOI: 10.1038/eye.2016.297.Latasiewicz M, Gourier H, Yusuf IH, et al. Hydroxychloroquine retinopathy: an emerging problem. Eye, 2017, 31(6): 972-976. DOI: 10.1038/eye.2016.297.
17、Marmor MF. Comparison of screening procedures in hydroxychloroquine toxicity. Arch Ophthalmol, 2012, 130(4): 461-469. DOI: 10.1001/archophthalmol.2011.371.Marmor MF. Comparison of screening procedures in hydroxychloroquine toxicity. Arch Ophthalmol, 2012, 130(4): 461-469. DOI: 10.1001/archophthalmol.2011.371.
18、Marmor MF, Kellner U, Lai TYY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016, 123(6): 1386-1394. DOI: 10.1016/j.ophtha.2016.01.058.Marmor MF, Kellner U, Lai TYY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016, 123(6): 1386-1394. DOI: 10.1016/j.ophtha.2016.01.058.
19、Kim KE, Kim YH, Kim J, et al. Macular ganglion cell complex and peripapillary retinal nerve fiber layer thicknesses in hydroxychloroquine retinopathy. Am J Ophthalmol. 2023, 245: 70-80. DOI: 10.1016/ j.ajo.2022.07.028.Kim KE, Kim YH, Kim J, et al. Macular ganglion cell complex and peripapillary retinal nerve fiber layer thicknesses in hydroxychloroquine retinopathy. Am J Ophthalmol. 2023, 245: 70-80. DOI: 10.1016/ j.ajo.2022.07.028.
20、Gil P, Nunes A, Farinha C, et al. Structural and functional characterization of the retinal effects of hydroxychloroquine treatment in healthy eyes. Ophthalmologica. 2019, 241(4): 226-233. DOI: 10.1159/000495308.Gil P, Nunes A, Farinha C, et al. Structural and functional characterization of the retinal effects of hydroxychloroquine treatment in healthy eyes. Ophthalmologica. 2019, 241(4): 226-233. DOI: 10.1159/000495308.
21、Pesch UEA, Fries JE, Bette S, et al. OPA1, the disease gene for autosomal dominant optic atrophy, is specifically expressed in ganglion cells and intrinsic neurons of the retina. Invest Ophthalmol Vis Sci. 2004, 45(11): 4217- 4225. DOI: 10.1167/iovs.03-1261.Pesch UEA, Fries JE, Bette S, et al. OPA1, the disease gene for autosomal dominant optic atrophy, is specifically expressed in ganglion cells and intrinsic neurons of the retina. Invest Ophthalmol Vis Sci. 2004, 45(11): 4217- 4225. DOI: 10.1167/iovs.03-1261.
22、KhosaS, KhanlouN, Khosa GS, et al. Hydroxychloroquine-induced autophagic vacuolar
myopathy with mitochondrial abnormalities. Neuropathology. 2018, 38(6): 646-652. DOI: 10.1111/neup.12520.KhosaS, KhanlouN, Khosa GS, et al. Hydroxychloroquine-induced autophagic vacuolar
myopathy with mitochondrial abnormalities. Neuropathology. 2018, 38(6): 646-652. DOI: 10.1111/neup.12520.